Diagonalizarea cuantică bazată pe eșantioane a unui Hamiltonian de chimie
Estimare de utilizare: sub un minut pe un procesor Heron r2 (NOTĂ: Aceasta este doar o estimare. Durata de rulare poate varia.)
Rezultate de învățare
După parcurgerea acestui tutorial, utilizatorii ar trebui să înțeleagă:
- Cum să folosești addon-ul SQD pentru Qiskit pentru a aproxima energia stării fundamentale a unui sistem molecular folosind șiruri de biți eșantionate de la o unitate de procesare cuantică (QPU).
- Cum să folosești ffsim pentru a construi un circuit ansatz local unitar cluster Jastrow (LUCJ) pentru simularea chimiei cuantice.
Condiții prealabile
Recomandăm utilizatorilor să se familiarizeze cu următoarele subiecte înainte de a parcurge acest tutorial:
- Chimie cuantică și a doua cuantificare
- Utilizarea primitivei Sampler pentru eșantionarea din circuite cuantice
Context
În acest tutorial, îți arătăm cum să post-procesezi eșantioane cuantice cu zgomot pentru a aproxima energia stării fundamentale a moleculei de azot la lungimea de echilibru a legăturii, utilizând addon-ul SQD pentru Qiskit pentru a implementa algoritmul de diagonalizare cuantică bazată pe eșantioane (SQD). Mai multe detalii despre software se găsesc în documentația corespunzătoare, inclusiv un exemplu simplu pentru a începe.
Acest tutorial este recomandat utilizatorilor familiarizați cu chimia cuantică: în special, familiarizarea cu găsirea energiilor stărilor fundamentale ale unei molecule. Pentru o prezentare detaliată a fluxului de lucru, consultă cursul despre algoritmul de diagonalizare cuantică.
SQD este o tehnică de găsire a valorilor proprii și a vectorilor proprii ai operatorilor cuantici, cum ar fi Hamiltonianul unui sistem cuantic, utilizând împreună calculul cuantic și calculul clasic distribuit. Calculul clasic distribuit este folosit pentru a procesa eșantioane obținute de la un procesor cuantic și pentru a proiecta și diagonaliza un Hamiltonian țintă într-un subspațiu pe care îl generează. Un flux de lucru bazat pe SQD are următorii pași:
- Alege un ansatz de circuit și aplică-l pe un calculator cuantic la o stare de referință (în acest caz, starea Hartree-Fock).
- Eșantionează șiruri de biți din starea cuantică rezultată.
- Rulează procedura de recuperare auto-consistentă a configurației pe șirurile de biți pentru a obține aproximarea stării fundamentale.
SQD funcționează bine atunci când starea proprie țintă este rară: funcția de undă este susținută de un set de stări de bază ale căror dimensiuni nu cresc exponențial cu dimensiunea problemei.
Chimie cuantică
Hamiltonianul unui sistem molecular poate fi scris ca
unde și sunt numere complexe numite integrale moleculare, care pot fi calculate din specificația moleculei folosind un program de calculator. În acest tutorial, calculăm integralele folosind pachetul software PySCF.
Pentru detalii despre derivarea Hamiltonianului molecular, consultă un manual de chimie cuantică (de exemplu, Modern Quantum Chemistry de Szabo și Ostlund). Pentru o explicație de nivel înalt a modului în care problemele de chimie cuantică sunt mapate pe calculatoare cuantice, urmărește prelegerea Mapping Problems to qubits de la Qiskit Global Summer School 2024.
Ansatzul local unitar cluster Jastrow (LUCJ)
SQD necesită un ansatz de circuit cuantic pentru a genera eșantioane. În acest tutorial, vom folosi ansatzul local unitar cluster Jastrow (LUCJ) datorită combinației dintre motivația fizică și compatibilitatea cu hardware-ul. Vom folosi ffsim pentru a construi circuitul ansatz.
Ansatzul LUCJ se adaptează la QPU-urile cu conectivitate limitată a qubiților. Orbitalele de spin sunt mapate pe qubiți astfel încât ansatzul nu necesită rutare cu porți SWAP. Hardware-ul IBM® are o topologie de qubiți cu rețea hex grea, caz în care putem adopta un model „zig-zag", reprezentat mai jos. În acest model, orbitalele cu același spin sunt mapate pe qubiți cu o topologie liniară (cercuri roșii și albastre), iar o conexiune între orbitale cu spin diferit există la fiecare al 4-lea orbital spațial, conexiunea fiind facilitată de un qubit ancilar (cercuri violet).
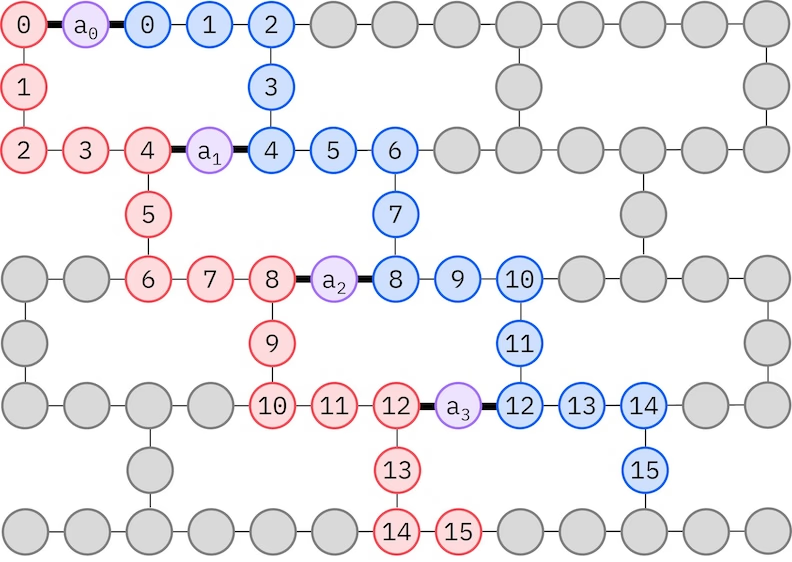
Recuperarea auto-consistentă a configurației
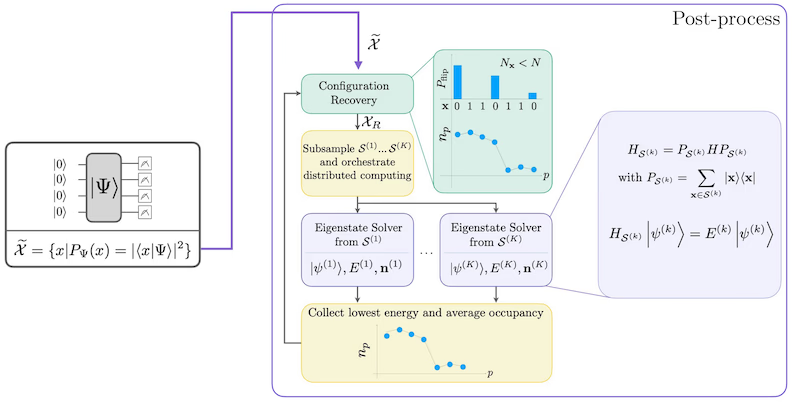
Procedura de recuperare auto-consistentă a configurației este concepută pentru a extrage cât mai mult semnal posibil din eșantioanele cuantice cu zgomot. Deoarece Hamiltonianul molecular conservă numărul de particule și spinul Z, are sens să alegi un ansatz de circuit care conservă și aceste simetrii. Când este aplicat la starea Hartree-Fock, starea rezultată are un număr fix de particule și spin Z în cazul fără zgomot. Prin urmare, jumătățile spin- și spin- ale oricărui șir de biți eșantionat din această stare ar trebui să aibă aceeași greutate Hamming ca în starea Hartree-Fock. Din cauza zgomotului din procesoarele cuantice actuale, unele șiruri de biți măsurate vor încălca această proprietate. O formă simplă de postselecție ar elimina aceste șiruri de biți, dar aceasta este risipitoare deoarece acele șiruri de biți ar putea conține totuși un anumit semnal. Procedura de recuperare auto-consistentă încearcă să recupereze o parte din acel semnal în post-procesare. Procedura este iterativă și necesită ca intrare o estimare a ocupanțelor medii ale fiecărui orbital în starea fundamentală, care este calculată mai întâi din eșantioanele brute. Procedura rulează în buclă, iar fiecare iterație are următorii pași:
- Pentru fiecare șir de biți care încalcă simetriile specificate, inversează biții printr-o procedură probabilistică concepută pentru a aduce șirul de biți mai aproape de estimarea curentă a ocupanțelor medii ale orbitalelor, pentru a obține un nou șir de biți.
- Colectează toate șirurile de biți vechi și noi care satisfac simetriile și eșantionează subseturi de dimensiune fixă, aleasă în prealabil.
- Pentru fiecare subset de șiruri de biți, proiectează Hamiltonianul în subspațiul generat de vectorii de bază corespunzători (consulta secțiunea anterioară pentru o descriere a acestor vectori de bază) și calculează o estimare a stării fundamentale a Hamiltonianului proiectat pe un calculator clasic.
- Actualizează estimarea ocupanțelor medii ale orbitalelor cu estimarea stării fundamentale cu cea mai mică energie.
Diagrama fluxului de lucru SQD
Fluxul de lucru SQD este reprezentat în diagrama următoare:
Cerințe
Înainte de a începe acest tutorial, asigură-te că ai instalat următoarele:
- Qiskit SDK v1.0 sau o versiune ulterioară, cu suport pentru vizualizare
- Qiskit Runtime v0.22 sau o versiune ulterioară (
pip install qiskit-ibm-runtime) - Addon-ul SQD pentru Qiskit v0.11 sau o versiune ulterioară (
pip install qiskit-addon-sqd) - ffsim v0.0.75 sau o versiune ulterioară (
pip install ffsim)
Configurare
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import math
import ffsim
import matplotlib.pyplot as plt
import numpy as np
import pyscf
import pyscf.cc
import pyscf.mcscf
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.primitives import StatevectorSampler
from qiskit.providers.fake_provider import GenericBackendV2
from qiskit_ibm_runtime import QiskitRuntimeService
from qiskit_ibm_runtime import SamplerV2 as Sampler
Exemplu la scară mică cu simulator
În acest tutorial, vom găsi o aproximare a stării fundamentale a unei molecule de azot în apropierea distanței de legătură de echilibru. Folosim mai întâi un set de baze STO-6G mic pentru a putea simula experimentul și a ne asigura că funcționează.
Pasul 1: Maparea intrărilor clasice la o problemă cuantică
Mai întâi, specificăm molecula și proprietățile sale.
# Specify molecule properties
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="sto-6g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Compute exact energy using FCI
reference_energy = cas.run().e_tot
print(f"norb = {norb}")
print(f"nelec = {nelec}")
converged SCF energy = -108.464957764796
CASCI E = -108.595987350986 E(CI) = -32.4115475088426 S^2 = 0.0000000
norb = 8
nelec = (5, 5)
Înainte de a construi Circuit-ul ansatz LUCJ, efectuăm mai întâi un calcul CCSD în celula de cod următoare. Amplitudinile și din acest calcul vor fi folosite pentru a inițializa parametrii ansatzului.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -108.5933309085008 E_corr = -0.1283731437052354
Acum, folosim ffsim pentru a crea Circuit-ul ansatz. Deoarece molecula noastră are o stare Hartree-Fock cu coajă închisă, folosim varianta echilibrată de spin a ansatzului UCJ, UCJOpSpinBalanced. Setăm optimize=True în metoda from_t_amplitudes pentru a activa factorizarea dublă „comprimată" a amplitudinilor (consulta The local unitary cluster Jastrow (LUCJ) ansatz din documentația ffsim pentru detalii).
Deoarece ansatzul LUCJ se adaptează la conectivitatea disponibilă a QPU, trebuie să inițializăm backend-ul QPU înainte de a crea ansatzul. Deocamdată, vom crea un backend generic cu o hartă de cuplare heavy-hex și un set de gate-uri la care ansatzul LUCJ se descompune natural. Apoi, vom folosi ffsim.qiskit.generate_lucj_pass_manager pentru a crea un pass manager specializat pentru transpilarea ansatzului LUCJ pe backend-ul dat conform layout-ului „zig-zag" descris în secțiunea de fundal despre ansatzul LUCJ. Această funcție utilizează o euristică de punctaj pentru a minimiza erorile asociate layout-ului selectat, ceea ce este important dacă backend-ul tău este un QPU real sau un simulator cu un model de zgomot. Pe lângă returnarea pass manager-ului, această funcție returnează și perechile de cuplare alfa-beta care pot fi implementate pe hardware. Dacă nu toate perechile pot fi implementate, emite un avertisment.
import warnings
from qiskit.transpiler import CouplingMap
warnings.formatwarning = lambda msg, *args, **kwargs: f"Warning: {msg}\n"
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
coupling_map = CouplingMap.from_heavy_hex(3)
backend = GenericBackendV2(
coupling_map.size(),
coupling_map=coupling_map,
basis_gates=["cp", "xx_plus_yy", "p", "x", "swap"],
)
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Pasul 2: Optimizează pentru execuția pe hardware cuantic
În continuare, optimizăm circuitul pentru un hardware țintă. De obicei, acest pas implică inițializarea backend-ului hardware și a unui pass manager pentru acel backend. Cu toate acestea, deoarece ansatzul LUCJ este adaptat la conectivitatea hardware-ului, am realizat deja aceste acțiuni în pasul anterior. Tot ce mai rămâne de făcut este să rulăm pass manager-ul pe circuit pentru a-l transpila într-un circuit ISA care poate fi executat direct pe QPU.
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
Gate counts: OrderedDict({'xx_plus_yy': 86, 'p': 16, 'measure': 16, 'cp': 15, 'x': 10, 'swap': 2, 'barrier': 1})
Pasul 3: Execuție folosind primitivele Qiskit
După optimizarea circuitului pentru execuția pe hardware, suntem pregătiți să îl rulăm pe hardware-ul țintă și să colectăm mostre pentru estimarea energiei stării fundamentale. Deoarece avem un singur circuit, vom folosi modul de execuție Job din Qiskit Runtime și vom executa circuitul nostru.
rng = np.random.default_rng()
sampler = StatevectorSampler(seed=rng)
job = sampler.run([isa_circuit], shots=100_000)
Warning: Trying to add QuantumRegister to a QuantumCircuit having a layout
primitive_result = job.result()
pub_result = primitive_result[0]
Pasul 4: Post-procesare și returnarea rezultatului în formatul clasic dorit
O metrică utilă pentru a evalua calitatea ieșirii QPU este numărul de configurații valide returnate. O configurație validă are numărul corect de particule și spin Z, ceea ce înseamnă că jumătatea dreaptă a șirului de biți are greutatea Hamming egală cu numărul de electroni cu spin-up, iar jumătatea stângă are greutatea Hamming egală cu numărul de electroni cu spin-down. Celula de mai jos calculează fracțiunea configurațiilor eșantionate care sunt valide.
def is_valid_bitstring(
bitstring: str, norb: int, nelec: tuple[int, int]
) -> bool:
n_alpha, n_beta = nelec
return (
len(bitstring) == 2 * norb
and bitstring[norb:].count("1") == n_alpha
and bitstring[:norb].count("1") == n_beta
)
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
Fraction of sampled configurations that are valid: 1.0
Toate șirurile de biți sunt valide deoarece eșantionăm circuitul pe un simulator fără zgomot. Când rulăm pe un QPU cu zgomot, fracțiunea va fi mai mică decât unu, dar sperăm că va fi mai mare decât fracțiunea la care ne-am aștepta dacă șirurile de biți ar fi eșantionate aleatoriu uniform, care este calculată în celula de mai jos.
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
Expected fraction of valid configurations from uniformly random bitstrings: 0.0478515625
Acum estimăm energia stării fundamentale a Hamiltonianului folosind funcția diagonalize_fermionic_hamiltonian. Această funcție realizează procedura de recuperare auto-consistentă a configurației pentru a rafina iterativ mostrele cuantice zgomotoase și a îmbunătăți estimarea energiei. Transmitem o funcție callback pentru a putea salva rezultatele intermediare în vederea analizei ulterioare. Consultă documentația API pentru explicații privind argumentele funcției diagonalize_fermionic_hamiltonian.
Aici, folosim argumentul initial_occupancies al funcției diagonalize_fermionic_hamiltonian pentru a specifica configurația Hartree-Fock ca estimare inițială pentru ocupanțele orbitale în starea fundamentală. Această abordare este rațională pentru sistemele în care starea fundamentală are sprijin semnificativ pe configurația Hartree-Fock, dar poate să nu fie adecvată în alte situații, deși metode computaționale mai avansate ar putea furniza estimări inițiale mai bune în acele cazuri. Specificarea initial_occupancies permite, de asemenea, recuperarea configurației să ruleze chiar dacă nu au fost eșantionate configurații valide, ceea ce poate fi cazul la eșantionarea unui circuit mare pe un QPU cu zgomot. Fără acest argument, recuperarea configurației ar eșua și ar genera o eroare dacă nu au fost furnizate configurații valide.
from functools import partial
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
solve_sci_batch,
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
Iteration 1
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Iteration 2
Subsample 0
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 1
Energy: -108.59275573641656
Subspace dimension: 900
Subsample 2
Energy: -108.59275573641656
Subspace dimension: 900
Final energy: -108.59275573641656
Final energy error: 0.0032316145694579745
Vizualizarea rezultatelor
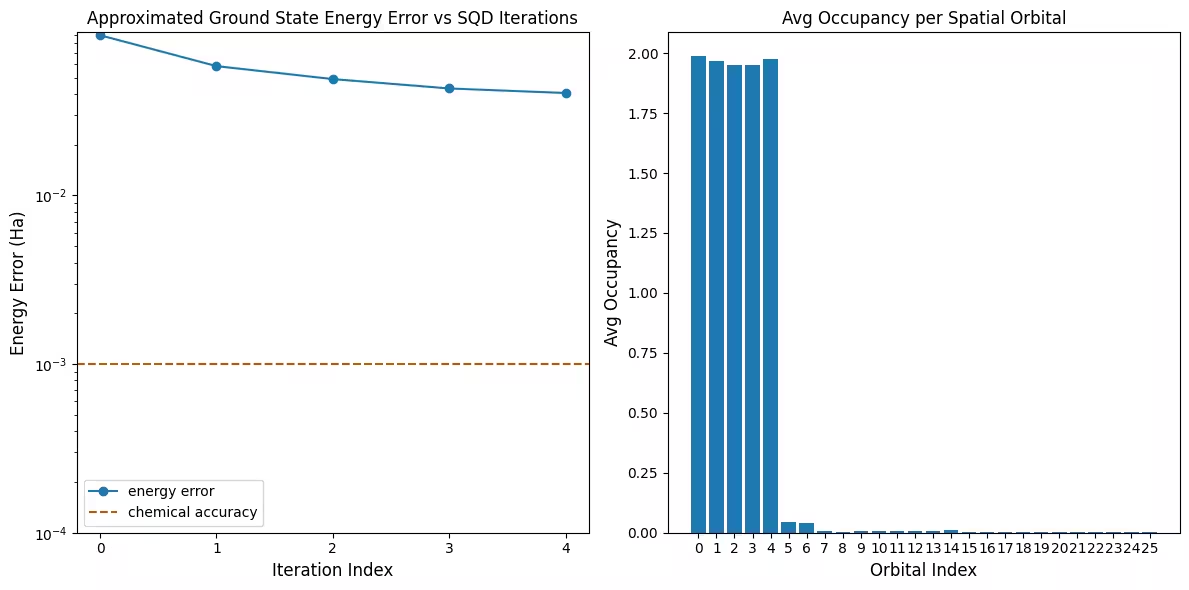
Primul grafic arată că în această simulare suntem deja în limita 1 mH față de răspunsul exact după prima iterație (precizia chimică este acceptată în general ca fiind 1 kcal/mol 1.6 mH). Acesta este totuși un sistem mic și, deoarece eșantioanele sunt fără zgomot, recuperarea configurației nu este necesară. Pe un sistem mai mare rulat pe un QPU cu zgomot, pot fi necesare mai multe iterații de recuperare a configurației, iar precizia finală poate fi mai slabă. În general, energia poate fi îmbunătățită permițând mai multe iterații de recuperare a configurației sau mărind numărul de mostre per lot.
Al doilea grafic arată ocupanța medie a fiecărei orbite spațiale după iterația finală. Putem observa că atât electronii cu spin-up, cât și cei cu spin-down ocupă primele cinci orbite cu probabilitate ridicată în soluțiile noastre.
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()

Exemplu la scară largă pe hardware real
Acum rulăm un exemplu mai mare pe hardware cuantic real. Aici, vom deriva un spațiu activ pentru molecula de azot din setul de baze cc-pVDZ.
Pașii 1-4
Aici reunim toți pașii într-un singur flux de lucru la scară mai mare, care este apoi rulat pe hardware cuantic real.
# ------------------------------ Step 1 ------------------------------
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="cc-pvdz",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
norb = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
n_alpha = (n_electrons + mol.spin) // 2
n_beta = (n_electrons - mol.spin) // 2
nelec = (n_alpha, n_beta)
cas = pyscf.mcscf.CASCI(scf, norb, nelec)
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), norb)
# Store reference energy from SCI calculation performed separately
reference_energy = -109.22802921665716
print(f"norb = {norb}")
print(f"nelec = {nelec}")
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(
scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]
).run()
t1 = ccsd.t1
t2 = ccsd.t2
# Set ansatz properties
n_reps = 1
pairs_aa = [(p, p + 1) for p in range(norb - 1)]
# Let generate_lucj_pass_manager determine the alpha-beta interactions
pairs_ab = None
# Initialize backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=133
)
print(f"Using backend {backend.name}")
# Create pass manager
pass_manager, pairs_ab = ffsim.qiskit.generate_lucj_pass_manager(
backend=backend,
norb=norb,
connectivity="heavy-hex",
interaction_pairs=(pairs_aa, pairs_ab),
optimization_level=3,
)
# Create the LUCJ ansatz operator
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(pairs_aa, pairs_ab),
# Setting optimize=True enables the "compressed" factorization
optimize=True,
# Limit the number of optimization iterations to prevent the code cell
# from running too long. Removing this line may improve results.
options=dict(maxiter=1000),
)
# create an empty quantum circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it
# to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(norb, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
# ------------------------------ Step 2 ------------------------------
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts: {isa_circuit.count_ops()}")
# ------------------------------ Step 3 ------------------------------
sampler = Sampler(mode=backend)
sampler.options.environment.job_tags = ["TUT_SQD"]
job = sampler.run([isa_circuit], shots=100_000)
primitive_result = job.result()
pub_result = primitive_result[0]
# ------------------------------ Step 4 ------------------------------
bit_array = pub_result.data.meas
num_valid = sum(
is_valid_bitstring(b, norb, nelec) for b in bit_array.get_bitstrings()
)
valid_fraction = num_valid / bit_array.num_shots
print(f"Fraction of sampled configurations that are valid: {valid_fraction}")
expected_fraction_random = (
math.comb(norb, n_alpha) * math.comb(norb, n_beta) / 2 ** (2 * norb)
)
print(
f"Expected fraction of valid configurations from uniformly random bitstrings: "
f"{expected_fraction_random}"
)
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 3
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Use the Hartree-Fock configuration as an initial guess for the
# orbital occupancies
initial_occupancies = (
np.array([1] * n_alpha + [0] * (norb - n_alpha)),
np.array([1] * n_beta + [0] * (norb - n_beta)),
)
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the
# sci_solver argument in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=norb,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
initial_occupancies=initial_occupancies,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
final_energy = result.energy + nuclear_repulsion_energy
energy_error = final_energy - reference_energy
print(f"Final energy: {final_energy}")
print(f"Final energy error: {energy_error}")
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - reference_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(
y=chem_accuracy,
color="#BF5700",
linestyle="--",
label="chemical accuracy",
)
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
plt.tight_layout()
plt.show()
converged SCF energy = -108.929838385609
norb = 26
nelec = (5, 5)
E(CCSD) = -109.2177884185544 E_corr = -0.2879500329450045
Using backend ibm_boston
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20), (24, 24)].
Removing interaction (24, 24) from the end.
Warning: Backend cannot accommodate pairs_ab=[(0, 0), (4, 4), (8, 8), (12, 12), (16, 16), (20, 20)].
Removing interaction (20, 20) from the end.
Gate counts: OrderedDict({'sx': 7039, 'rz': 6990, 'cz': 1858, 'x': 61, 'measure': 52, 'barrier': 1})
Fraction of sampled configurations that are valid: 0.02124
Expected fraction of valid configurations from uniformly random bitstrings: 9.607888706852918e-07
Iteration 1
Subsample 0
Energy: -109.13889134249762
Subspace dimension: 120409
Subsample 1
Energy: -109.11785470455858
Subspace dimension: 110889
Subsample 2
Energy: -109.13234360554011
Subspace dimension: 130321
Iteration 2
Subsample 0
Energy: -109.16392179579177
Subspace dimension: 223729
Subsample 1
Energy: -109.16281938332986
Subspace dimension: 223729
Subsample 2
Energy: -109.16955816711932
Subspace dimension: 233289
Iteration 3
Subsample 0
Energy: -109.17905772999075
Subspace dimension: 324900
Subsample 1
Energy: -109.17532445048462
Subspace dimension: 357604
Subsample 2
Energy: -109.1733168689756
Subspace dimension: 348100
Iteration 4
Subsample 0
Energy: -109.18437778820451
Subspace dimension: 474721
Subsample 1
Energy: -109.18450164209159
Subspace dimension: 476100
Subsample 2
Energy: -109.18493571190754
Subspace dimension: 487204
Iteration 5
Subsample 0
Energy: -109.18616522497996
Subspace dimension: 622521
Subsample 1
Energy: -109.18652868888333
Subspace dimension: 644809
Subsample 2
Energy: -109.18753326484406
Subspace dimension: 585225
Final energy: -109.18753326484406
Final energy error: 0.040495951813099396
Pași următori
Dacă ți s-a părut interesant acest material, s-ar putea să fii interesat de următoarele resurse:
- Diagonalizarea cuantică Krylov bazată pe eșantioane a unui model de rețea fermionic - un tutorial înrudit care folosește circuite de evoluție temporală în loc de un ansatz variațional
- Extinde fluxurile de lucru SQD de chimie cu soluționatorul Dice - o pagină care arată cum să folosești software-ul Dice mai eficient pentru diagonalizare
- Documentația API addon-ului SQD - referință pentru funcția
diagonalize_fermionic_hamiltonian - Chemistry beyond the scale of exact diagonalization on a quantum-centric supercomputer - articolul pe care se bazează acest tutorial