Diagonalizarea cuantică Krylov bazată pe eșantionare a unui model de rețea fermionică
Estimare de utilizare: Nouă secunde pe un procesor Heron r2 (NOTĂ: Aceasta este doar o estimare. Timpul tău de execuție poate varia.)
Rezultate ale învățării
După parcurgerea acestui tutorial, utilizatorii ar trebui să înțeleagă:
- Cum să folosească addon-ul SQD pentru Qiskit pentru a aproxima energia stării fundamentale a unui model de rețea folosind șiruri de biți eșantionate de pe o unitate de procesare cuantică (QPU).
- Cum să folosească ffsim pentru a construi circuite de evoluție temporală pentru simulare fermionică.
- Cum să combine eșantioane din mai multe circuite pentru post-procesare cu algoritmul de diagonalizare Krylov bazată pe eșantionare (SKQD).
Cerințe prealabile
Sugerăm utilizatorilor să fie familiarizați cu următoarele subiecte înainte de a parcurge acest tutorial:
- Diagonalizarea cuantică bazată pe eșantionare a unui Hamiltonian chimic
- Diagonalizarea cuantică Krylov a Hamiltonienilor de rețea
- Primitive Qiskit
Fundal
Acest tutorial arată cum să folosești diagonalizarea cuantică bazată pe eșantionare (SQD) pentru a estima energia stării fundamentale a unui model de rețea fermionică. În mod concret, studiem modelul Anderson unidimensional cu o singură impuritate (SIAM), care este utilizat pentru a descrie impuritățile magnetice încorporate în metale.
Acest tutorial urmează un flux de lucru similar cu tutorialul înrudit Diagonalizarea cuantică bazată pe eșantionare a unui Hamiltonian chimic. Totuși, o diferență cheie constă în modul în care sunt construite circuitele cuantice. Celălalt tutorial folosește un ansatz variațional euristic, atrăgător pentru Hamiltonieni chimici cu potențial milioane de termeni de interacțiune. Pe de altă parte, acest tutorial folosește circuite care aproximează evoluția temporală guvernată de Hamiltonian. Astfel de circuite pot fi adânci, ceea ce face ca această abordare să fie mai potrivită pentru aplicații la modele de rețea. Vectorii de stare pregătiți de aceste circuite formează baza unui subspațiu Krylov, iar ca rezultat, algoritmul converge în mod demonstrabil și eficient spre starea fundamentală, în condiții potrivite.
Abordarea utilizată în acest tutorial poate fi privită ca o combinație a tehnicilor folosite în SQD și diagonalizarea cuantică Krylov (KQD). Abordarea combinată este uneori denumită diagonalizare cuantică Krylov bazată pe eșantionare (SQKD). Consultă Diagonalizarea cuantică Krylov a Hamiltonienilor de rețea pentru un tutorial despre metoda KQD.
Acest tutorial se bazează pe lucrarea "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization", la care poți apela pentru mai multe detalii.
Modelul Anderson cu o singură impuritate (SIAM)
Hamiltonianul SIAM unidimensional este suma a trei termeni:
unde
Aici, sunt operatorii fermionici de creare/anihilare pentru al sit de baie cu spin , sunt operatorii de creare/anihilare pentru modul de impuritate, iar . , și sunt numere reale care descriu interacțiunile de hopping, on-site și hibridizare, iar este un număr real care specifică potențialul chimic.
Observă că Hamiltonianul este o instanță specifică a Hamiltonianului generic de electroni în interacțiune,
unde constă din termeni cu un corp, care sunt cuadratici în operatorii de creare și anihilare fermionici, iar constă din termeni cu două corpuri, care sunt quartici. Pentru SIAM,
iar conține restul termenilor din Hamiltonian. Pentru a reprezenta Hamiltonianul în mod programatic, stocăm matricea și tensorul .
Bazele de poziție și impuls
Din cauza simetriei de translație aproximative din , nu ne așteptăm ca starea fundamentală să fie rară în baza de poziție (baza orbitalilor în care este specificat Hamiltonianul de mai sus). Performanța SQD este garantată doar dacă starea fundamentală este rară, adică are pondere semnificativă pe doar un număr mic de stări ale bazei de calcul. Pentru a îmbunătăți raritatea stării fundamentale, efectuăm simularea în baza orbitalilor în care este diagonal. Numim această bază baza de impuls. Deoarece este un Hamiltonian fermion cuadratic, el poate fi diagonalizat eficient printr-o rotație orbitală.
Evoluția temporală aproximativă guvernată de Hamiltonian
Pentru a aproxima evoluția temporală guvernată de Hamiltonian, folosim o descompunere Trotter-Suzuki de ordinul doi,
Sub transformarea Jordan-Wigner, evoluția temporală prin corespunde unei singure porți CPhase între orbitalii de spin-up și spin-down de la situl de impuritate. Deoarece este un Hamiltonian fermion cuadratic, evoluția temporală prin corespunde unei rotații orbitale.
Stările bazei Krylov , unde este dimensiunea subspațiului Krylov, sunt formate prin aplicarea repetată a unui singur pas Trotter, astfel
În fluxul de lucru bazat pe SQD de mai jos, vom eșantiona din acest set de circuite și vom post-procesa setul combinat de șiruri de biți cu SQD. Această abordare contrastează cu cea utilizată în tutorialul înrudit Diagonalizarea cuantică bazată pe eșantionare a unui Hamiltonian chimic, unde eșantioanele erau extrase dintr-un singur circuit variațional euristic.
Cerințe
Înainte de a începe acest tutorial, asigură-te că ai instalate următoarele:
- Qiskit SDK v1.0 sau mai recent, cu suport de vizualizare
- Qiskit Runtime v0.22 sau mai recent (
pip install qiskit-ibm-runtime) - SQD Qiskit addon v0.11 sau mai recent (
pip install qiskit-addon-sqd) - ffsim v0.0.72 sau mai recent (
pip install ffsim)
Exemplu la scară mică cu simulator
Pasul 1: Maparea problemei pe un Circuit cuantic
Mai întâi, generăm Hamiltonianul SIAM în baza de poziție. Hamiltonianul este reprezentat de matricea și tensorul . Apoi îl rotim în baza de impuls. În baza de poziție, plasăm impuritatea la primul sit. Totuși, când rotim în baza de impuls, mutăm impuritatea la un sit central pentru a facilita interacțiunile cu alți orbitali.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime scipy
import numpy as np
import pyscf.fci
def siam_hamiltonian(
norb: int,
hopping: float,
onsite: float,
hybridization: float,
chemical_potential: float,
) -> tuple[np.ndarray, np.ndarray]:
"""Hamiltonian for the single-impurity Anderson model."""
# Place the impurity on the first site
impurity_orb = 0
# One body matrix elements in the "position" basis
h1e = np.zeros((norb, norb))
np.fill_diagonal(h1e[:, 1:], -hopping)
np.fill_diagonal(h1e[1:, :], -hopping)
h1e[impurity_orb, impurity_orb + 1] = -hybridization
h1e[impurity_orb + 1, impurity_orb] = -hybridization
h1e[impurity_orb, impurity_orb] = chemical_potential
# Two body matrix elements in the "position" basis
h2e = np.zeros((norb, norb, norb, norb))
h2e[impurity_orb, impurity_orb, impurity_orb, impurity_orb] = onsite
return h1e, h2e
def momentum_basis(norb: int) -> np.ndarray:
"""Get the orbital rotation to change from the position to the momentum basis."""
n_bath = norb - 1
# Orbital rotation that diagonalizes the bath (non-interacting system)
hopping_matrix = np.zeros((n_bath, n_bath))
np.fill_diagonal(hopping_matrix[:, 1:], -1)
np.fill_diagonal(hopping_matrix[1:, :], -1)
_, vecs = np.linalg.eigh(hopping_matrix)
# Expand to include impurity
orbital_rotation = np.zeros((norb, norb))
# Impurity is on the first site
orbital_rotation[0, 0] = 1
orbital_rotation[1:, 1:] = vecs
# Move the impurity to the center
new_index = n_bath // 2
perm = np.r_[1 : (new_index + 1), 0, (new_index + 1) : norb]
orbital_rotation = orbital_rotation[:, perm]
return orbital_rotation
def rotated(
h1e: np.ndarray, h2e: np.ndarray, orbital_rotation: np.ndarray
) -> tuple[np.ndarray, np.ndarray]:
"""Rotate the orbital basis of a Hamiltonian."""
h1e_rotated = np.einsum(
"ab,Aa,Bb->AB",
h1e,
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
h2e_rotated = np.einsum(
"abcd,Aa,Bb,Cc,Dd->ABCD",
h2e,
orbital_rotation,
orbital_rotation.conj(),
orbital_rotation,
orbital_rotation.conj(),
optimize="greedy",
)
return h1e_rotated, h2e_rotated
# Total number of spatial orbitals, including the bath sites and the impurity
# This should be an even number
norb = 8
# System is half-filled
nelec = (norb // 2, norb // 2)
# One orbital is the impurity, the rest are bath sites
n_bath = norb - 1
# Hamiltonian parameters
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian in position basis
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
# Rotate to momentum basis
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
# In the momentum basis, the impurity is placed in the center
impurity_index = n_bath // 2
# Use PySCF to compute the exact ground state energy
reference_energy, _ = pyscf.fci.direct_spin1.kernel(h1e, h2e, norb, nelec)
from typing import Sequence
import ffsim
import scipy
from qiskit import QuantumCircuit, QuantumRegister
from qiskit.circuit import CircuitInstruction, Qubit
from qiskit.circuit.library import CPhaseGate, XGate, XXPlusYYGate
def prepare_initial_state(qubits: Sequence[Qubit], norb: int, nocc: int):
"""Prepare initial state."""
assert norb >= 8
x_gate = XGate()
rot = XXPlusYYGate(0.5 * np.pi, -0.5 * np.pi)
for i in range(nocc):
yield CircuitInstruction(x_gate, [qubits[i]])
yield CircuitInstruction(x_gate, [qubits[norb + i]])
for i in range(3):
for j in range(nocc - i - 1, nocc + i, 2):
yield CircuitInstruction(rot, [qubits[j], qubits[j + 1]])
yield CircuitInstruction(
rot, [qubits[norb + j], qubits[norb + j + 1]]
)
yield CircuitInstruction(rot, [qubits[j + 1], qubits[j + 2]])
yield CircuitInstruction(
rot, [qubits[norb + j + 1], qubits[norb + j + 2]]
)
def trotter_step(
qubits: Sequence[Qubit],
time_step: float,
one_body_evolution: np.ndarray,
h2e: np.ndarray,
impurity_index: int,
norb: int,
):
"""A Trotter step."""
# Assume the two-body interaction is just the on-site interaction of the impurity
onsite = h2e[
impurity_index, impurity_index, impurity_index, impurity_index
]
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# One-body evolution for the full time
yield CircuitInstruction(
ffsim.qiskit.OrbitalRotationJW(norb, one_body_evolution), qubits
)
# Two-body evolution for half the time
yield CircuitInstruction(
CPhaseGate(-0.5 * time_step * onsite),
[qubits[impurity_index], qubits[norb + impurity_index]],
)
# Time step
time_step = 0.2
# Number of Krylov basis states
krylov_dim = 8
# Initialize circuit
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
# Generate initial state
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
# Create list of circuits, starting with the initial state circuit
circuits = [circuit.copy()]
# Add time evolution circuits to the list
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
# Remove measurements
circuit.remove_final_measurements()
# Append another Trotter step
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
# Measure qubits
circuit.measure_all()
# Add a copy of the circuit to the list
circuits.append(circuit.copy())
În continuare, generăm circuitele pentru a produce stările bazei Krylov. Pentru fiecare specie de spin, starea inițială este dată de superpoziția tuturor excitațiilor posibile ale celor trei electroni mai apropiați de nivelul Fermi în cele 4 moduri goale cele mai apropiate, pornind de la starea , realizată prin aplicarea a șapte porți XXPlusYYGate. Stările evoluate în timp sunt produse prin aplicări succesive ale unui pas Trotter de ordinul doi.
Pentru o descriere mai detaliată a acestui model și a modului în care sunt proiectate circuitele, consultă "Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization".
circuits[0].draw("mpl", scale=0.4, fold=-1)
circuits[-1].draw("mpl", scale=0.4, fold=-1)
Pasul 2: Optimizează problema pentru execuție cuantică
În continuare, optimizăm circuitul pentru un hardware țintă. Deocamdată, vom crea un backend generic cu un număr specificat de qubiți și un set de porți la care circuitele de evoluție temporală se descompun în mod natural.
from qiskit.providers.fake_provider import GenericBackendV2
backend = GenericBackendV2(
2 * norb, basis_gates=["cp", "xx_plus_yy", "p", "x"]
)
Acum folosim Qiskit pentru a transpila circuitele către backend-ul țintă.
from qiskit.transpiler import generate_preset_pass_manager
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
Pasul 3: Execută folosind primitivele Qiskit
După optimizarea circuitelor pentru execuție hardware, suntem gata să le rulăm pe hardware-ul țintă și să colectăm eșantioane pentru estimarea energiei stării fundamentale. După ce folosim primitiva Sampler pentru a eșantiona șiruri de biți din fiecare circuit, combinăm toate rezultatele într-un singur dicționar de numărători și reprezentăm grafic cele mai frecvente 20 de șiruri de biți eșantionate.
from qiskit.visualization import plot_histogram
from qiskit.primitives import StatevectorSampler
# Sample from the circuits
sampler = StatevectorSampler()
job = sampler.run(isa_circuits, shots=500)
from qiskit.primitives import BitArray
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
plot_histogram(bit_array.get_counts(), number_to_keep=20)
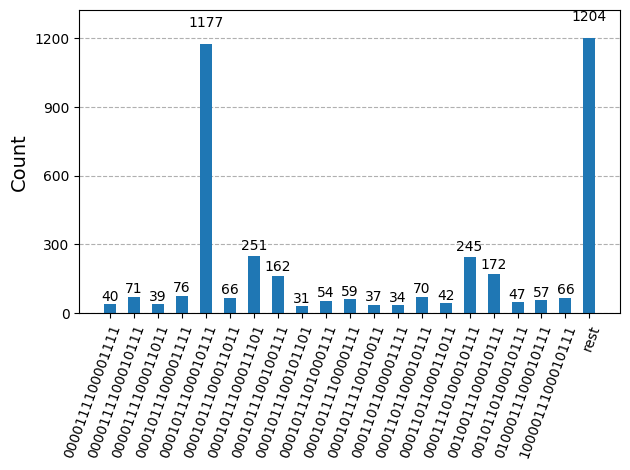
Pasul 4: Post-procesează și returnează rezultatul în formatul clasic dorit
Acum rulăm algoritmul SQD folosind funcția diagonalize_fermionic_hamiltonian. Consultă documentația API pentru explicații privind argumentele acestei funcții.
from qiskit_addon_sqd.fermion import (
SCIResult,
diagonalize_fermionic_hamiltonian,
)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -13.4222953188441
Subspace dimension: 529
Subsample 1
Energy: -13.42237556285828
Subspace dimension: 784
Subsample 2
Energy: -13.422045397387413
Subspace dimension: 529
Iteration 2
Subsample 0
Energy: -13.422379583305478
Subspace dimension: 900
Subsample 1
Energy: -13.422376197704326
Subspace dimension: 841
Subsample 2
Energy: -13.422421162849295
Subspace dimension: 1089
Iteration 3
Subsample 0
Energy: -13.422421164670345
Subspace dimension: 1156
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421205869572
Subspace dimension: 1156
Iteration 4
Subsample 0
Energy: -13.422421494558726
Subspace dimension: 1225
Subsample 1
Energy: -13.422421492737689
Subspace dimension: 1156
Subsample 2
Energy: -13.422421492737689
Subspace dimension: 1156
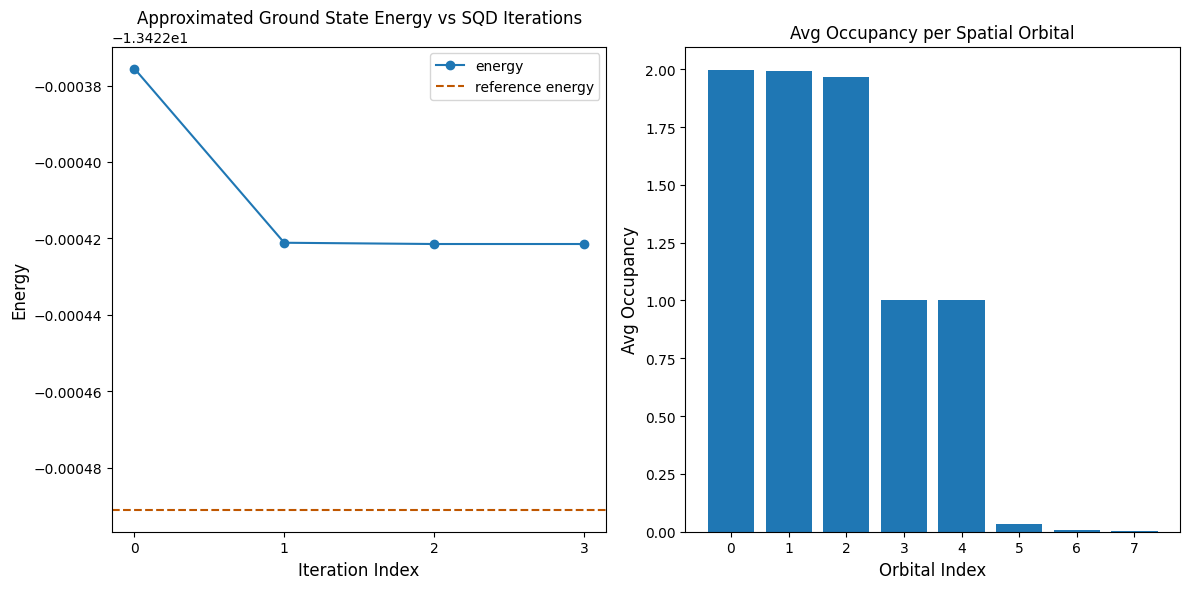
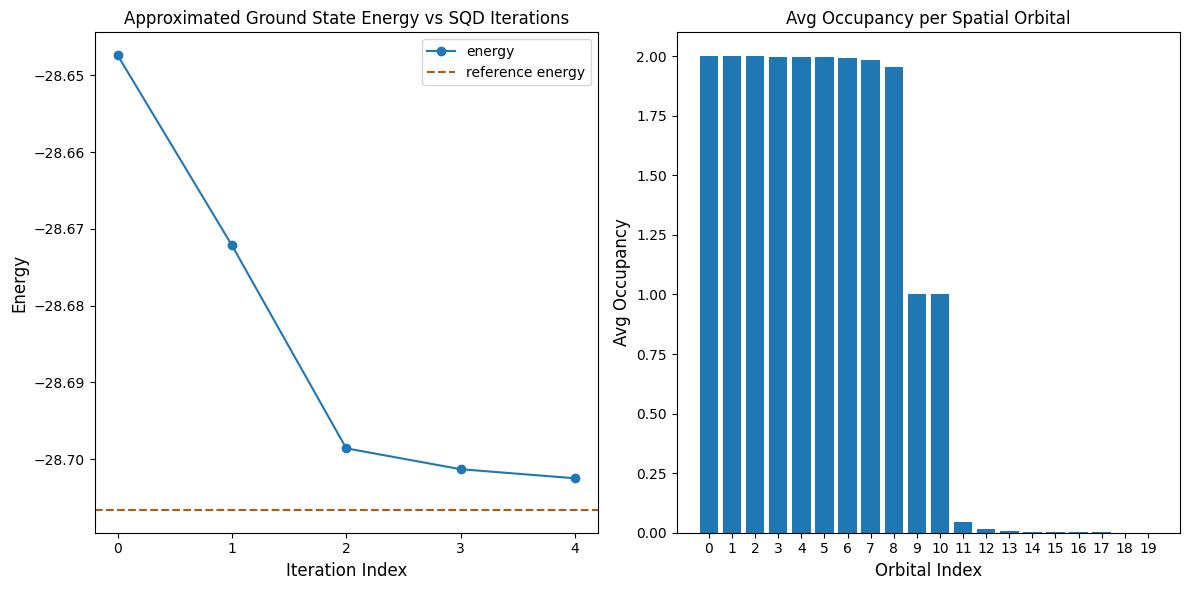
Următoarea celulă de cod reprezintă grafic rezultatele. Primul grafic arată energia calculată în funcție de numărul de iterații de recuperare a configurației, iar al doilea grafic arată ocuparea medie a fiecărui orbital spațial după iterația finală. Deoarece este o problemă atât de mică, prima iterație ne aduce deja foarte aproape de energia exactă (observați scala axei y).
import matplotlib.pyplot as plt
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
# Data for energies plot
x1 = range(len(result_history))
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Reference energy: -13.42249
SQD energy: -13.42242
Absolute error: 0.00007
Verifică energia
Energia returnată de SQD este garantată să fie o limită superioară a energiei adevărate a stării fundamentale. Valoarea energiei poate fi verificată deoarece SQD returnează și coeficienții vectorului de stare care aproximează starea fundamentală. Poți calcula energia din vectorul de stare folosind matricele de densitate redusă de ordinul unu și doi, după cum este demonstrat în celula de cod de mai jos.
rdm1 = result.sci_state.rdm(rank=1, spin_summed=True)
rdm2 = result.sci_state.rdm(rank=2, spin_summed=True)
energy = np.sum(h1e_momentum * rdm1) + 0.5 * np.sum(h2e_momentum * rdm2)
print(f"Recomputed energy: {energy:.5f}")
Recomputed energy: -13.42242
Exemplu la scară mare cu hardware
Acum rulăm un exemplu mai mare pe un QPU real. Pentru energia de referință, folosim rezultatele unui calcul DMRG efectuat separat.
from qiskit_ibm_runtime import SamplerV2 as Sampler
from qiskit_ibm_runtime import QiskitRuntimeService
# Model parameters
norb = 20
nelec = (norb // 2, norb // 2)
n_bath = norb - 1
hybridization = 1.0
hopping = 1.0
onsite = 10.0
chemical_potential = -0.5 * onsite
# Generate Hamiltonian and orbital rotation
h1e, h2e = siam_hamiltonian(
norb=norb,
hopping=hopping,
onsite=onsite,
hybridization=hybridization,
chemical_potential=chemical_potential,
)
orbital_rotation = momentum_basis(norb)
h1e_momentum, h2e_momentum = rotated(h1e, h2e, orbital_rotation.T.conj())
impurity_index = n_bath // 2
# Set reference energy to DMRG value computed separately
reference_energy = -28.70659686
# Algorithm parameters
time_step = 0.2
krylov_dim = 8
# Construct circuits
qubits = QuantumRegister(2 * norb, name="q")
circuit = QuantumCircuit(qubits)
for instruction in prepare_initial_state(qubits, norb=norb, nocc=norb // 2):
circuit.append(instruction)
circuit.measure_all()
circuits = [circuit.copy()]
one_body_evolution = scipy.linalg.expm(-1j * time_step * h1e_momentum)
for i in range(krylov_dim - 1):
circuit.remove_final_measurements()
for instruction in trotter_step(
qubits,
time_step,
one_body_evolution,
h2e_momentum,
impurity_index,
norb,
):
circuit.append(instruction)
circuit.measure_all()
circuits.append(circuit.copy())
# Initialize hardware backend
service = QiskitRuntimeService()
backend = service.least_busy(
operational=True, simulator=False, min_num_qubits=127
)
print(f"Using backend {backend.name}")
# Transpile to backend
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend
)
isa_circuits = pass_manager.run(circuits)
# Sample from the circuits
sampler = Sampler(backend)
sampler.options.environment.job_tags = ["TUT_SKQD"]
job = sampler.run(isa_circuits, shots=500)
# Combine the shots from the individual Trotter circuits
bit_array = BitArray.concatenate_shots(
[result.data.meas for result in job.result()]
)
# Run configuration recovery and diagonalization
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy}")
print(
f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}"
)
rng = np.random.default_rng(24)
result = diagonalize_fermionic_hamiltonian(
h1e_momentum,
h2e_momentum,
bit_array,
samples_per_batch=100,
norb=norb,
nelec=nelec,
num_batches=3,
max_iterations=5,
symmetrize_spin=True,
callback=callback,
seed=rng,
)
# Plot results
min_es = [
min(result, key=lambda res: res.energy).energy
for result in result_history
]
min_id, min_e = min(enumerate(min_es), key=lambda x: x[1])
x1 = range(len(result_history))
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
axs[0].plot(x1, min_es, label="energy", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].axhline(
y=reference_energy,
color="#BF5700",
linestyle="--",
label="reference energy",
)
axs[0].set_title("Approximated Ground State Energy vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy", fontdict={"fontsize": 12})
axs[0].legend()
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Reference energy: {reference_energy:.5f}")
print(f"SQD energy: {min_e:.5f}")
print(f"Absolute error: {abs(min_e - reference_energy):.5f}")
plt.tight_layout()
plt.show()
Using backend ibm_boston
Iteration 1
Subsample 0
Energy: -28.63965951544449
Subspace dimension: 9801
Subsample 1
Energy: -28.625588929202006
Subspace dimension: 9409
Subsample 2
Energy: -28.647371834135498
Subspace dimension: 8281
Iteration 2
Subsample 0
Energy: -28.67213260849567
Subspace dimension: 29584
Subsample 1
Energy: -28.670340686158816
Subspace dimension: 27225
Subsample 2
Energy: -28.669976379525988
Subspace dimension: 31329
Iteration 3
Subsample 0
Energy: -28.68622875601382
Subspace dimension: 36100
Subsample 1
Energy: -28.698569623143126
Subspace dimension: 34225
Subsample 2
Energy: -28.694848533971882
Subspace dimension: 33856
Iteration 4
Subsample 0
Energy: -28.69883392844593
Subspace dimension: 42025
Subsample 1
Energy: -28.701289495200996
Subspace dimension: 38025
Subsample 2
Energy: -28.699319594978245
Subspace dimension: 45369
Iteration 5
Subsample 0
Energy: -28.701936886834154
Subspace dimension: 51076
Subsample 1
Energy: -28.702468711812013
Subspace dimension: 53824
Subsample 2
Energy: -28.702298147575938
Subspace dimension: 52900
Reference energy: -28.70660
SQD energy: -28.70247
Absolute error: 0.00413
Pași următori
Dacă ai găsit această lucrare interesantă, s-ar putea să fii interesat de următoarele materiale:
- Diagonalizarea cuantică bazată pe eșantionare a unui Hamiltonian chimic - un tutorial înrudit care utilizează un ansatz variațional euristic în loc de circuite Trotter
- Diagonalizarea cuantică Krylov a Hamiltonienilor de rețea - un tutorial despre metoda KQD
- Documentația API a addon-ului SQD - referință pentru funcția
diagonalize_fermionic_hamiltonian - Quantum-Centric Algorithm for Sample-Based Krylov Diagonalization - lucrarea pe care se bazează acest tutorial