HI-VQE Chemistry - O Funcție Qiskit de Qunova Computing
# Added by doQumentation — required packages for this notebook
!pip install -q qiskit-ibm-catalog qiskit-ibm-runtime
# This cell is hidden from users
from qiskit_ibm_runtime import QiskitRuntimeService
service = QiskitRuntimeService()
instance = service.active_account()["instance"]
backend_name = service.least_busy(operational=True, min_num_qubits=16).name
Consultă referința API
Funcțiile Qiskit sunt o funcționalitate experimentală disponibilă numai utilizatorilor IBM Quantum® Premium Plan, Flex Plan și On-Prem (prin IBM Quantum Platform API) Plan. Acestea se află în stadiu de previzualizare și pot suferi modificări.
Package versions
The code on this page was developed using the following requirements. We recommend using these versions or newer.
qiskit-ibm-runtime~=0.45.0
Prezentare generală
În chimia cuantică, problema structurii electronice se concentrează pe găsirea soluțiilor ecuației Schrödinger electronice — funcțiile de undă cuantice care descriu comportamentul electronilor sistemului. Aceste funcții de undă sunt vectori de amplitudini complexe, fiecare amplitudine corespunzând contribuției unei configurații electronice posibile.
Starea fundamentală este funcția de undă cu cea mai mică energie a sistemului și are o importanță deosebită în studiul sistemelor moleculare. Abordarea cea mai precisă pentru calcularea stării fundamentale ia în considerare toate configurațiile electronice posibile, însă aceasta devine imposibil de gestionat pentru sisteme mai mari, deoarece numărul configurațiilor crește exponențial cu dimensiunea sistemului.
Handover Iterative Variational Quantum Eigensolver (HI-VQE) este o metodă hibridă cuantic-clasică inovatoare pentru estimarea precisă a stării fundamentale a sistemelor moleculare. Integrează hardware cuantic cu calcul clasic, utilizând procesoare cuantice pentru a explora eficient configurațiile electronice candidate și calculând funcția de undă rezultantă pe calculatoare clasice. Prin generarea de funcții de undă compacte, dar precise din punct de vedere chimic, HI-VQE îmbunătățește cercetarea și descoperirile în chimia cuantică și știința materialelor.

HI-VQE reduce complexitatea computațională a problemei structurii electronice prin estimarea eficientă a stării fundamentale cu o precizie ridicată. Se concentrează pe un subset atent selectat al celor mai relevante configurații electronice, optimizând atât precizia, cât și eficiența.
Combinând punctele forte ale calculatoarelor clasice și cuantice, HI-VQE rafinează și îmbunătățește iterativ estimarea curentă a funcției de undă. Tehnicile sale unice de construire a subspatiului ajută la eficientizarea selecției configurațiilor, astfel încât utilizatorii să aibă un control computational mai mare și o precizie îmbunătățită în simulările de chimie cuantică.
Dacă dorești să înveți mai în profunzime despre algoritm, poți citi lucrarea de cercetare asociată.
Descriere
Numărul de configurații electronice pentru un sistem molecular crește exponențial cu dimensiunea sistemului. Cu toate acestea, pentru anumite stări electronice, cum ar fi starea fundamentală, este obișnuit ca doar o mică fracțiune din configurații să contribuie semnificativ la energia stării. Metodele de interacțiune de configurație selectată (SCI) exploatează această sparsitate pentru a reduce costurile computaționale prin identificarea și concentrarea pe cele mai relevante configurații. Acest subset de configurații este denumit subspațiu.
HI-VQE valorifică eficiența inerentă a calculatoarelor cuantice pentru reprezentarea sistemelor moleculare în scopul facilitării căutării în subspațiu. Integrează subrutine clasice și cuantice pentru a rezolva problema structurii electronice cu o precizie ridicată. Spre deosebire de metodele SCI cuantice existente, HI-VQE combină antrenamentul variațional, construirea iterativă a subspațiului și filtrarea configurațiilor prin pre-diagonalizare pentru a îmbunătăți eficiența prin reducerea măsurătorilor cuantice, a iterațiilor și a costurilor de diagonalizare clasică. HI-VQE poate fi astfel aplicat unor sisteme moleculare mai mari care necesită mai mulți Qubiți și reduce costul rezolvării unei probleme de o dimensiune dată la același grad de precizie.
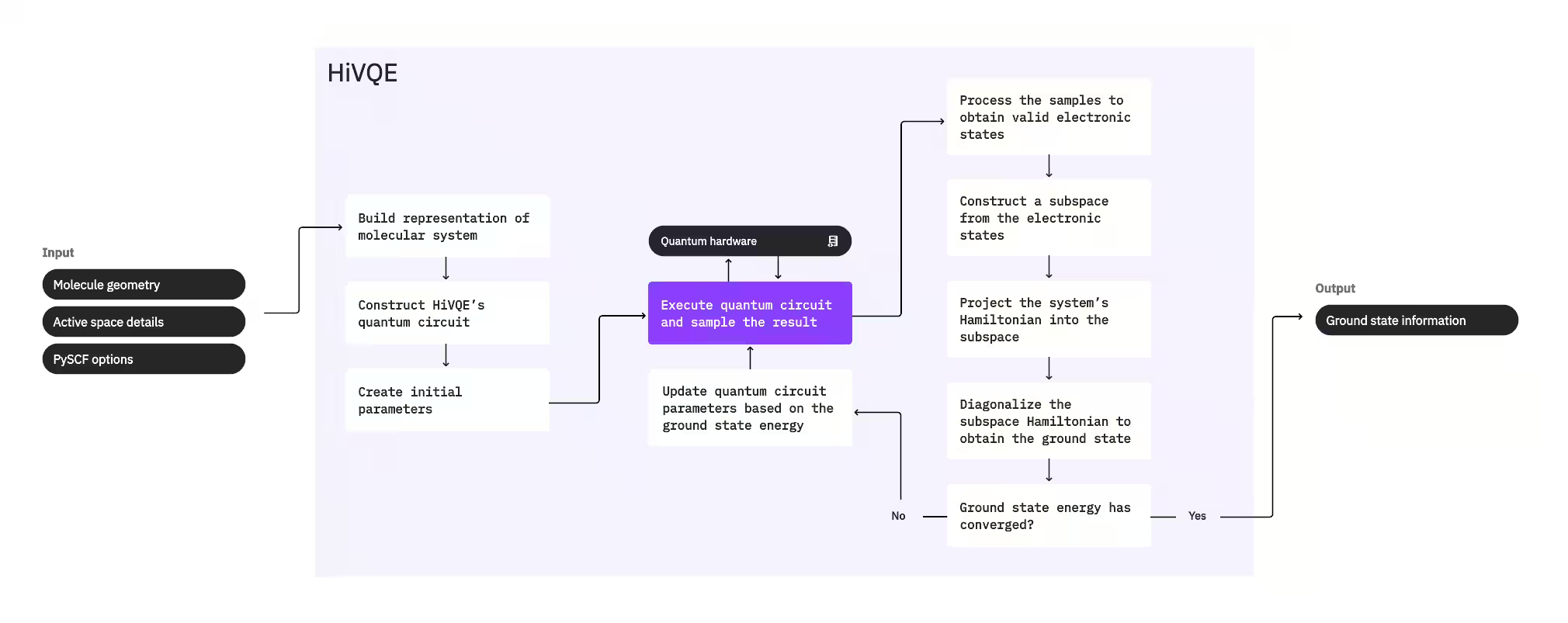
Pentru a calcula starea fundamentală a unui sistem, HI-VQE utilizează mai întâi pachetul clasic de chimie PySCF pentru a genera o reprezentare moleculară din datele de intrare furnizate de utilizator, cum ar fi geometria moleculară și alte informații moleculare. Intră apoi într-o buclă de optimizare hibridă cuantic-clasică, rafinând iterativ un subspațiu pentru a reprezenta optim starea fundamentală, minimizând în același timp numărul de configurații incluse. Bucla continuă până când sunt îndeplinite criterii de convergență, cum ar fi dimensiunea subspațiului sau stabilitatea energiei, după care sunt furnizate funcția de undă a stării fundamentale calculate și energia. Aceste rezultate pot fi utilizate pentru a construi suprafețe de energie potențială precise și pentru a efectua analize suplimentare ale sistemului.
Bucla de optimizare se concentrează pe ajustarea parametrilor unui Circuit cuantic pentru a genera un subspațiu de înaltă calitate. HI-VQE oferă trei opțiuni de circuit cuantic: excitation_preserving, efficient_su2 și LUCJ. Optimizarea este inițializată aproape de starea de referință Hartree-Fock datorită adecvabilității sale generale. Circuit-ul este apoi executat pe un dispozitiv cuantic și configurațiile sunt eșantionate din starea cuantică rezultantă înainte de a fi returnate ca șiruri binare. Datorită zgomotului dispozitivului cuantic, unele configurații eșantionate pot fi invalide din punct de vedere fizic, neconservând numărul de electroni sau spinul. HI-VQE abordează aceasta folosind procesul de recuperare a configurațiilor din pachetul qiskit-addon-sqd, astfel încât utilizatorii pot fie să corecteze configurațiile invalide, fie să le elimine.
Configurațiile valide parcurg apoi un pas opțional de filtrare pentru a le elimina pe cele estimate să contribuie minim. Aceasta reduce dimensiunea subspațiului, scăzând astfel costul pasului de diagonalizare. Dacă filtrarea este activată, atunci se construiește un Hamiltonian de subspațiu preliminar din configurațiile valide și se efectuează o diagonalizare cu criterii de terminare foarte relaxate. Deși precizia amplitudinilor rezultante pentru fiecare configurație este scăzută, aceasta este eficientă pentru a prezice ce configurații să fie excluse din subspațiu în această iterație și este rapidă de calculat.
Configurațiile selectate sunt adăugate la subspațiu, iar Hamiltonianul sistemului este proiectat în acest subspațiu. Subspațiul se actualizează iterativ, păstrând cele mai relevante configurații de-a lungul iterațiilor. Această abordare contrastează cu metodele alternative deoarece Circuit-ul cuantic nu trebuie să aproximeze întreaga stare fundamentală la fiecare pas.
Ulterior, Hamiltonianul de subspațiu este diagonalizat clasic pentru a obține cea mai mică valoare proprie și vectorul propriu corespunzător, reprezentând o aproximare a stării fundamentale și a energiei sale. Pe măsură ce calitatea subspațiului se îmbunătățește prin iterații, starea fundamentală calculată aproximează mai bine starea fundamentală reală. Un pas suplimentar de filtrare poate fi efectuat în acest punct pentru a elimina din subspațiu orice configurații care nu contribuie substanțial la starea fundamentală calculată. Acest pas asigură că subspațiul transferat în iterația următoare este cât mai compact posibil. Aceasta este evaluată pe baza amplitudinilor returnate de diagonalizare, deoarece acestea reprezintă contribuția de importanță a fiecărei configurații la starea fundamentală calculată.
O verificare a convergenței determină apoi dacă antrenamentul suplimentar ar îmbunătăți rezultatele. Dacă da, se efectuează un pas opțional de expansiune clasică, parametrii Circuit-ului cuantic sunt actualizați pentru a minimiza în continuare energia calculată și procesul se repetă. Pasul de expansiune clasică generează configurații suplimentare pentru subspațiu, completând configurațiile eșantionate de pe dispozitivul cuantic. Identifică mai întâi configurația cu amplitudinea cea mai mare în rezultatele diagonalizării, înainte de a genera noi configurații cu excitații simple și duble din configurația identificată. Numărul dorit din aceste configurații este apoi adăugat la subspațiu.
Odată ce s-a determinat că iterațiile au convergat, HI-VQE returnează starea fundamentală calculată (sub forma stărilor din subspațiu și a amplitudinilor lor în funcția de undă a stării fundamentale), energia sa și o măsură a varianței energiei care indică dacă starea calculată formează o stare proprie a Hamiltonianului sistemului.
Utilizatorii pot decide Circuit-ul cuantic utilizat și numărul de shot-uri efectuate pentru fiecare circuit cuantic, precum și controla dimensiunea subspațiului sau activa generarea clasică de configurații suplimentare pentru a asista configurațiile generate cuantic. În acest fel, utilizatorii pot adapta comportamentul HI-VQE pentru aplicațiile dorite.
Licențiere
Te rog să reții că utilizarea acestei Funcții Qiskit este limitată la probleme care necesită cel mult 20 de qubiți, dacă nu se obține o licență care acordă o limită mai mare.
Te rog să trimiți un e-mail la qiskit.support@qunovacomputing.com dacă dorești să faci o solicitare privind obținerea unei licențe.
Noțiuni introductive
Mai întâi, solicită acces la funcție. Apoi, autentifică-te folosind cheia ta API IBM Quantum® și, presupunând că ți-ai salvat deja contul în mediul local, selectează Funcția Qiskit după cum urmează:
import reprlib
from qiskit_ibm_catalog import QiskitFunctionsCatalog
catalog = QiskitFunctionsCatalog(channel="ibm_quantum_platform")
function = catalog.load("qunova/hivqe-chemistry")
Exemplu
Primul exemplu arată cum se calculează energia stării fundamentale pentru o moleculă de NH3 folosind algoritmul HI-VQE.
Definirea geometriei moleculare și a opțiunilor
Geometria moleculară a NH3 este furnizată cu coordonate carteziene separate prin ";" pentru fiecare atom.
# Define the molecule geometry
geometry = """
N -0.85188 -0.02741 0.03141;
H 0.16545 0.00593 -0.01648;
H -1.16348 -0.39357 -0.86702;
H -1.16348 0.94228 0.06281;
"""
Opțiuni suplimentare pot fi definite și furnizate pentru sistemul molecular în formatul de dicționar următor.
# Configure some options for the job.
molecule_options = {"basis": "sto3g"}
hivqe_options = {"shots": 100, "max_iter": 20}
Execută funcția cu intrările de geometrie și opțiuni.
# Run HI-VQE
job = function.run(
geometry=geometry,
# `backend_name` is the name of a backend with at least 16 qubits,
# for example, "ibm_marrakesh".
backend_name=backend_name,
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
Este o idee bună să afișezi ID-ul jobului funcției, astfel încât să poată fi furnizat în solicitările de suport dacă ceva merge greșit.
print("Job ID:", job.job_id)
Job ID: e5ced6f2-fd1d-4244-a6aa-bd27cfb0cdee
Acest exemplu utilizează apoi 16 qubiți cu 8 orbitali ai bazei sto3g pentru o moleculă de NH3. Verifică starea sau returnează rezultatele workload-ului tău Qiskit Function după cum urmează:
print(job.status())
QUEUED
După finalizarea jobului, rezultatele pot fi obținute cu instanța result().
result = job.result()
# Output can be long, so we display a shortened representation
shortened_result = reprlib.repr(result)
print(shortened_result)
{'eigenvector': [0.9824448589364075, 0.009527106392132133, 6.854074372058527e-08, 3.591500190038039e-07, 0.0012975231577544268, 2.310159709002111e-05, ...], 'energy': -55.52108557170985, 'energy_history': [-55.51901898989887, -55.52056881448526, -55.52065046778772, -55.520690696813716, -55.520691108428, -55.520708448092634, ...], 'energy_variance': 3.066239097617371e-10, ...}
Pentru a accesa energia stării fundamentale, folosește cheia "energy". Cheia "eigenvector" furnizează coeficienții CI cu notația bitstring corespunzătoare a configurației electronice stocate cu "states" din rezultate.
fci_energy = -55.521148034704126 # the exact energy using FCI method
hivqe_energy = result["energy"]
print(
f"|Exact Energy - HI-VQE Energy|: "
f"{abs(fci_energy - hivqe_energy) * 1000} mHa"
)
print(f"Sampled Number of States: {len(result['states'])}")
|Exact Energy - HI-VQE Energy|: 0.06246299427914437 mHa
Sampled Number of States: 1936
Performanță
Această secțiune prezintă calculele de referință demonstrate ale HI-VQE pentru un caz cu 24 de qubiți pentru Li2S, un caz cu 40 de qubiți pentru o moleculă N2 și un caz cu 44 de qubiți pentru un sistem FeP-NO.
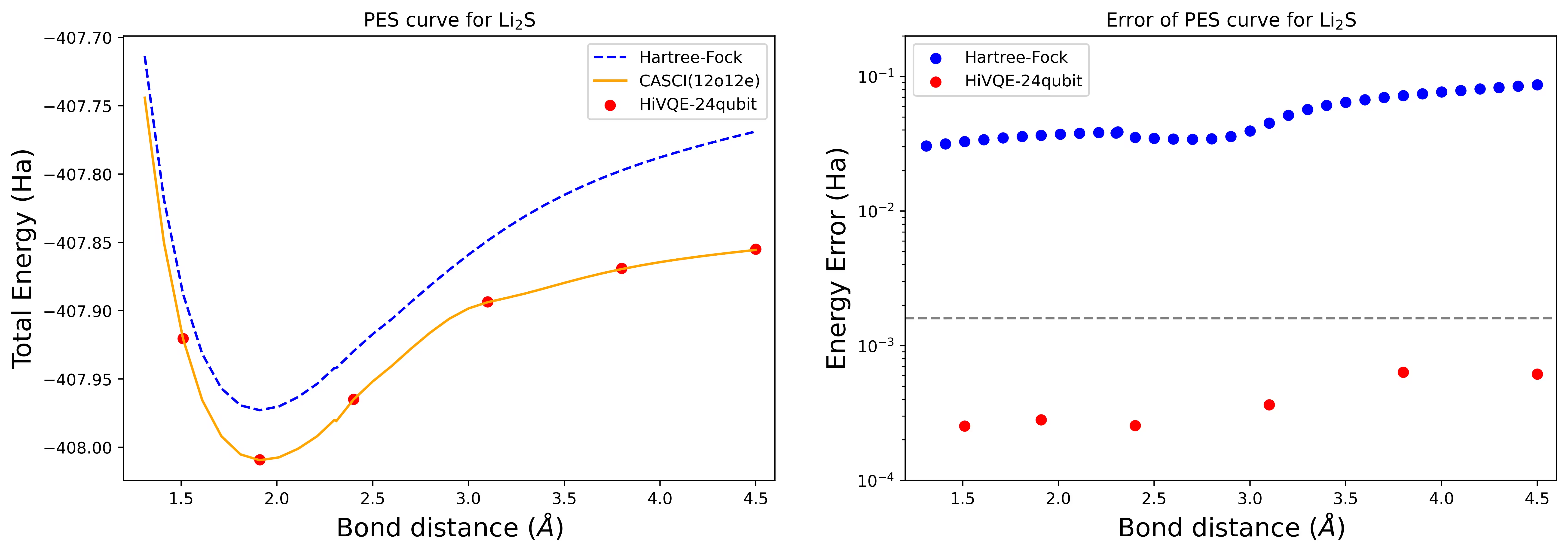
Curba suprafeței de energie potențială de disociere pentru o moleculă Li2S cu 24 de qubiți
Curba PES este prezentată cu referința FCI și estimarea inițială din RHF, împreună cu eroarea de energie față de referința FCI.
 .
.
Calculele au fost efectuate cu următoarele geometrii și opțiuni.
# This cell is hidden from users
backend_name = service.least_busy(operational=True, min_num_qubits=38).name
# Define Li2S geometries
Li2S_geoms = {
"Li2S_1.51": "S -1.239044 0.671232 -0.030374;Li -1.506327 0.432403 -1.498949;Li -0.899996 0.973348 1.826768;",
"Li2S_2.40": "S -1.741432 0.680397 0.346702;Li -0.529307 0.488006 -1.729343;Li -1.284307 0.989409 2.177209;",
"Li2S_3.80": "S -2.707255 0.674298 0.909161;Li 0.079218 0.552012 -1.671656;Li -0.927010 0.931502 1.557063;",
}
# Configure some options for the job.
molecule_options = {
"basis": "sto3g",
}
hivqe_options = {
"shots": 100,
"max_iter": 20,
}
results = []
for geom in ["Li2S_1.51", "Li2S_2.40", "Li2S_3.80"]:
# Run HI-VQE
job = function.run(
geometry=Li2S_geoms[geom],
backend_name=backend_name, # can use any device with at least 38 qubits
max_states=2000,
max_expansion_states=10,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
results.append(job.result())
Punctele roșii reprezintă rezultatele calculului HI-VQE pentru șase geometrii diferite, iar trei geometrii corespunzătoare valorilor de 1,51, 2,40 și 3,80 Angstromi sunt furnizate ca intrare în celula de mai sus.
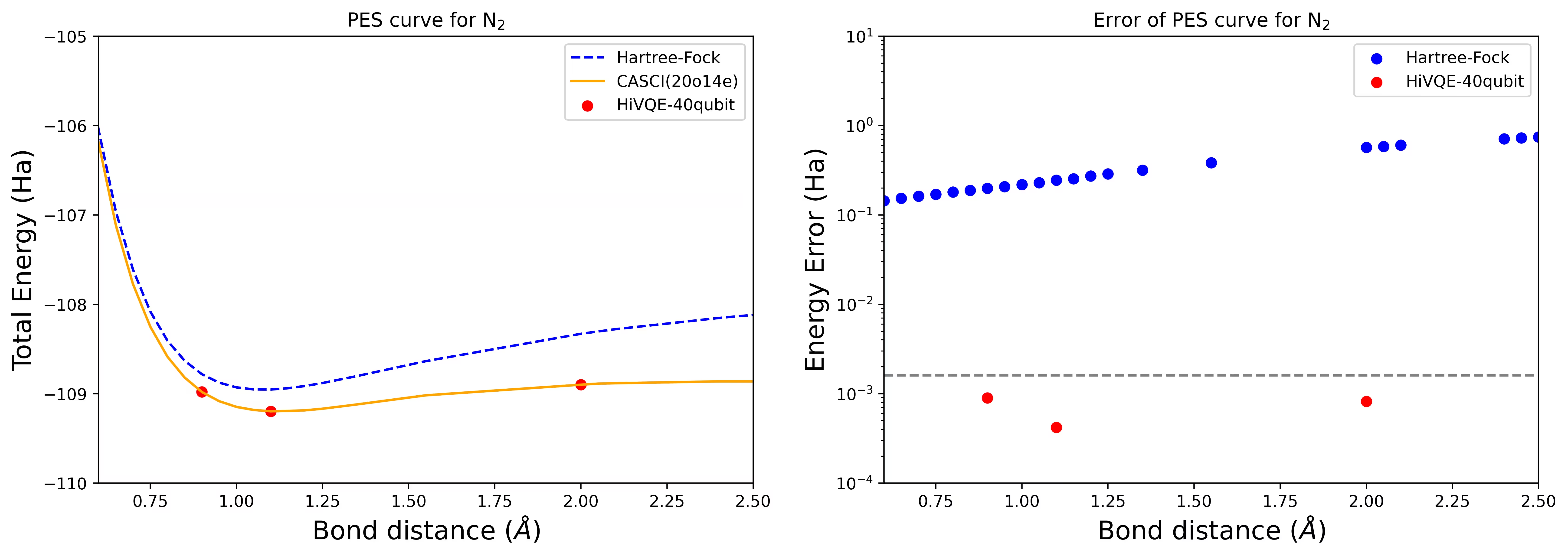
Curba PES de disociere pentru o moleculă N2 cu 40 de qubiți
Molecula de azot a fost identificată ca un sistem multireference cu contribuții mari de energie de corelație dincolo de starea Hartree-Fock. Am efectuat un calcul de referință pentru molecula N2 cu baza cc-pvdz, (20o,14e) folosind selecția orbitală activă homo-lumo. Numărul spațiului activ complet (CAS) pentru a reprezenta această problemă este 6.009.350.400. Nu este posibil să obții soluția problemei de valori proprii (pentru energie și structură electronică) cu acest număr de stări folosind un desktop performant (16cpu/64GB). Cu HI-VQE, poți căuta eficient subspațiul stărilor CAS pentru a găsi rezultate cu acuratețe chimică, economisind semnificativ resursele de calcul. Graficele următoare arată curba PES a calculului HI-VQE cu 40 de qubiți pentru disocierea moleculei N2.
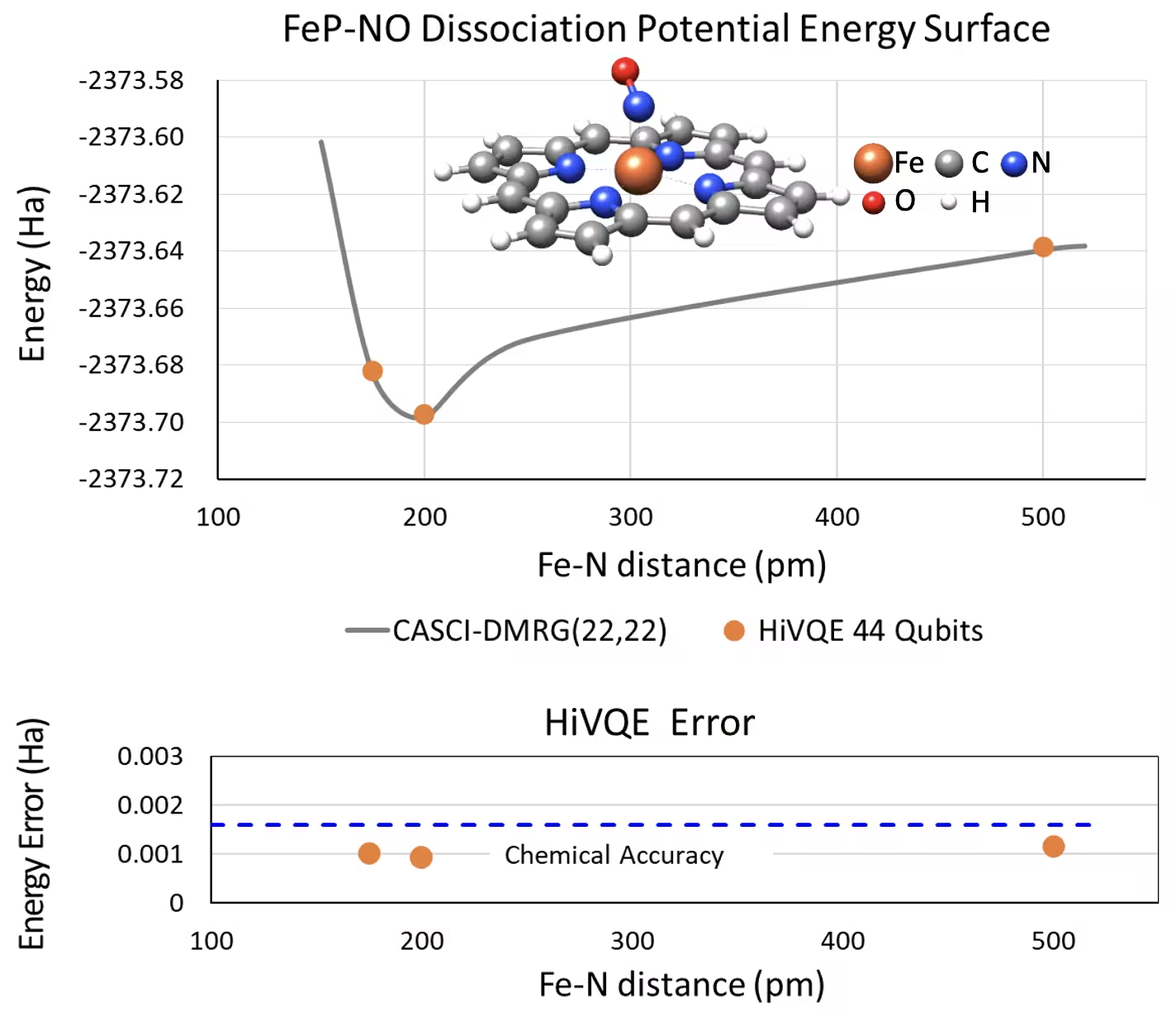
Curba PES de disociere pentru fier(II)-porfirina penta-coordonată cu un sistem NO cu 44 de qubiți
Un alt sistem chimic interesant este complexul fier(II)-porfirina (FeP) cu un ligand de oxid nitric (NO) coordonat, care reprezintă un sistem metaloporfirinic relevant biologic ce joacă roluri esențiale în diverse procese fiziologice. În acest exemplu, HI-VQE a fost utilizat pentru a estima curba exactă a suprafeței de energie potențială a interacțiunii intermoleculare dintre FeP și NO (energia stării fundamentale pentru geometrii separate diferit). Sistemul combinat are 450 de orbitali și 202 de electroni (450o,202e) cu baza 6-31g(d) în total. Selecția orbitală activă homo-lumo a fost utilizată pentru a calcula cazul mai mic față de cazul real cu (22o,22e). Din rezultatele de referință următoare, am reușit să atingem acuratețea chimică (> 1,6 mHa) față de un calcul de chimie cu calculator clasic de ultimă generație CASCI(DMRG) (22o,22e) de referință.
Valori de referință
- Dimensiunea exactă a matricei reprezintă numărul de determinanți pentru soluția exactă, cum ar fi FCI și CASCI.
- Calculul HI-VQE eșantionează și calculează subspațiul acesteia (adică dimensiunea matricei HI-VQE).
- Timpul total include timpul de execuție QPU și rulările Qiskit Function cu CPU.
- Acuratețea este estimată din diferența de energie față de soluția exactă.
| Sistem chimic | Număr de qubiți | Dimensiunea exactă a matricei | Dimensiunea matricei HI-VQE | E(diff) față de exact (mHa) | Număr de iterații | Timp total | Utilizare timp de execuție QPU |
|---|---|---|---|---|---|---|---|
| (8o,10e) | 16 | 3136 | 1936 | 0,08 | 6 | 37 s | 34 s |
| (10o,10e) | 20 | 63504 | 3969 | 0,60 | 5 | 250 s | 50 s |
| (15o,10e) | 30 | 9018009 | 49729 | 0,90 | 5 | 354 s | 54 s |
| (16o,14e) | 32 | 130873600 | 1798281 | 1,10 | 9 | 6531 s | 121 s |
| (18o,24e) | 36 | 344622096 | 399424 | 0,90 | 24 | 5174 s | 130 s |
| (20o,14e) | 40 | 6009350400 | 9012004 | 1,20 | 21 | 46547 s | 258 s |
Preluarea mesajelor de eroare
Dacă sarcina ta de lucru eșuează, starea va fi ERROR și apelarea job.result() va genera o excepție:
job = function.run(
geometry="invalid-geometry", # This will cause an error
backend_name=backend_name,
max_states=2000,
max_expansion_states=15,
molecule_options=molecule_options,
hivqe_options=hivqe_options,
)
job.result()
job.status()
'ERROR'
Obține asistență
Poți trimite un e-mail la qiskit.support@qunovacomputing.com pentru ajutor cu această funcție.
Dacă dorești ajutor pentru depanarea unei erori specifice, te rog să furnizezi ID-ul jobului Function al jobului care a întâmpinat eroarea.
Pași următori
- Solicită acces la funcție completând acest formular.
- Vizitează referința API pentru această Funcție Qiskit.
- Încearcă tutorialul Calculează curba PES de disociere pentru FeP-NO cu HI-VQE.
- Consultă Pellow-Jarman, A., et al. (2025). HIVQE: Handover Iterative Variational Quantum Eigensolver for Efficient Quantum Chemistry Calculations. arXiv preprint arXiv:2503.06292.
- Încearcă tutorialul Curbe PES de disociere cu Qunova HiVQE.