Îmbunătățirea estimării energiei unui Hamiltonian de chimie cu SQD
În acest tutorial implementăm un pattern Qiskit care arată cum să post-procesezi eșantioane cuantice zgomotoase pentru a găsi o aproximare a stării fundamentale a unui Hamiltonian de chimie: molecula la echilibru în baza 6-31G. Vom urma o abordare de diagonalizare cuantică bazată pe eșantionare pentru a procesa eșantioane preluate dintr-un ansatz de circuit cuantic cu 36 de Qubiți (în acest caz, un circuit LUCJ). Pentru a ține cont de efectul zgomotului cuantic, se folosește tehnica de recuperare a configurației.
Patternul poate fi descris în patru pași:
- Pasul 1: Maparea la problema cuantică
- Generează un ansatz pentru estimarea stării fundamentale
- Pasul 2: Optimizarea problemei
- Transpilează ansatz-ul pentru Backend
- Pasul 3: Executarea experimentelor
- Extrage eșantioane din ansatz folosind primitiva
Sampler
- Extrage eșantioane din ansatz folosind primitiva
- Pasul 4: Post-procesarea rezultatelor
- Bucla de recuperare auto-consistentă a configurației
- Post-procesează întregul set de eșantioane de tip bitstring, folosind cunoașterea prealabilă a numărului de particule și a ocupanței orbitale medii calculată la cea mai recentă iterație.
- Creează probabilistic loturi de subeșantioane din bitstring-uri recuperate.
- Proiectează și diagonalizează Hamiltonianul molecular peste fiecare subspațiu eșantionat.
- Salvează energia minimă a stării fundamentale găsită în toate loturile și actualizează ocupanța orbitală medie.
- Bucla de recuperare auto-consistentă a configurației
Pentru acest exemplu, Hamiltonianul de electroni în interacțiune are forma generică:
/ sunt operatorii fermonici de creare/anihilare asociați celui de-al -lea element al bazei și spinului . și sunt integralele electronice de un corp și, respectiv, de două corpuri.
Fluxul de lucru SQD cu recuperare auto-consistentă a configurației este ilustrat în diagrama de mai jos.
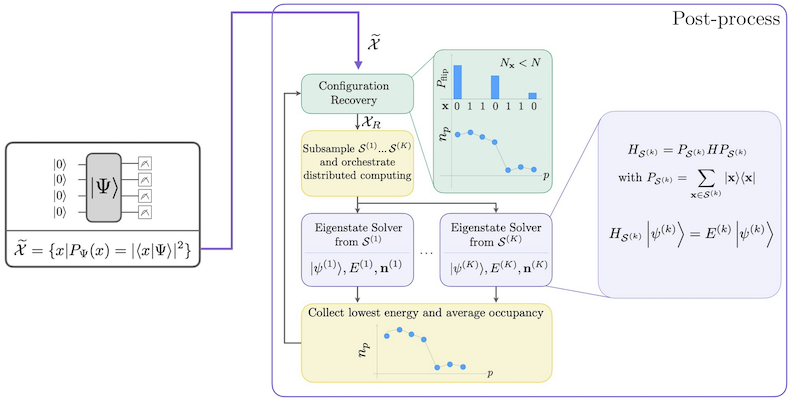
SQD funcționează bine atunci când starea proprie țintă este rară: funcția de undă este susținută într-un set de stări de bază ale cărei dimensiuni nu cresc exponențial cu dimensiunea problemei. În acest scenariu, diagonalizarea Hamiltonianului proiectat în subspațiul definit de :
produce o bună aproximare a stării proprii țintă. Rolul dispozitivului cuantic este de a produce eșantioane ale membrilor din . Mai întâi, un circuit cuantic pregătește starea pe dispozitivul cuantic. Se folosește codificarea Jordan-Wigner. În consecință, membrii bazei de calcul reprezintă stări Fock (configurații/determinanți electronici). Circuitul este eșantionat în baza de calcul, obținând setul de configurații zgomotoase . Configurațiile sunt reprezentate prin bitstring-uri. Setul este apoi transmis blocului de post-procesare clasic, unde se folosește tehnica de recuperare auto-consistentă a configurației. În cadrul SQD, rolul dispozitivului cuantic este de a produce o distribuție de probabilitate.
Pasul 1: Maparea problemei la un circuit cuantic
În acest tutorial, vom aproxima energia stării fundamentale a unei molecule de . Mai întâi, vom specifica molecula și proprietățile sale. Apoi, vom crea un ansatz local unitary cluster Jastrow (LUCJ) (Circuit cuantic) pentru a genera eșantioane de pe un calculator cuantic în scopul estimării energiei stării fundamentale.
Mai întâi, vom specifica molecula și proprietățile sale.
# Added by doQumentation — required packages for this notebook
!pip install -q ffsim matplotlib numpy pyscf qiskit qiskit-addon-sqd qiskit-ibm-runtime
import warnings
warnings.filterwarnings("ignore")
import pyscf
import pyscf.cc
import pyscf.mcscf
# Specify molecule properties
open_shell = False
spin_sq = 0
# Build N2 molecule
mol = pyscf.gto.Mole()
mol.build(
atom=[["N", (0, 0, 0)], ["N", (1.0, 0, 0)]],
basis="6-31g",
symmetry="Dooh",
)
# Define active space
n_frozen = 2
active_space = range(n_frozen, mol.nao_nr())
# Get molecular integrals
scf = pyscf.scf.RHF(mol).run()
num_orbitals = len(active_space)
n_electrons = int(sum(scf.mo_occ[active_space]))
num_elec_a = (n_electrons + mol.spin) // 2
num_elec_b = (n_electrons - mol.spin) // 2
cas = pyscf.mcscf.CASCI(scf, num_orbitals, (num_elec_a, num_elec_b))
mo = cas.sort_mo(active_space, base=0)
hcore, nuclear_repulsion_energy = cas.get_h1cas(mo)
eri = pyscf.ao2mo.restore(1, cas.get_h2cas(mo), num_orbitals)
# Compute exact energy
exact_energy = cas.run().e_tot
converged SCF energy = -108.835236570775
CASCI E = -109.046671778080 E(CI) = -32.8155692383188 S^2 = 0.0000000
Apoi, vom crea ansatz-ul. Ansatz-ul LUCJ este un circuit cuantic parametrizat și îl vom inițializa cu amplitudinile t2 și t1 obținute dintr-un calcul CCSD.
# Get CCSD t2 amplitudes for initializing the ansatz
ccsd = pyscf.cc.CCSD(scf, frozen=[i for i in range(mol.nao_nr()) if i not in active_space]).run()
t1 = ccsd.t1
t2 = ccsd.t2
E(CCSD) = -109.0398256929734 E_corr = -0.2045891221988311
Vom folosi pachetul ffsim pentru a crea și inițializa ansatz-ul cu amplitudinile t2 și t1 calculate mai sus. Deoarece molecula noastră are o stare Hartree-Fock cu înveliș închis, vom folosi varianta echilibrată de spin a ansatz-ului UCJ, UCJOpSpinBalanced.
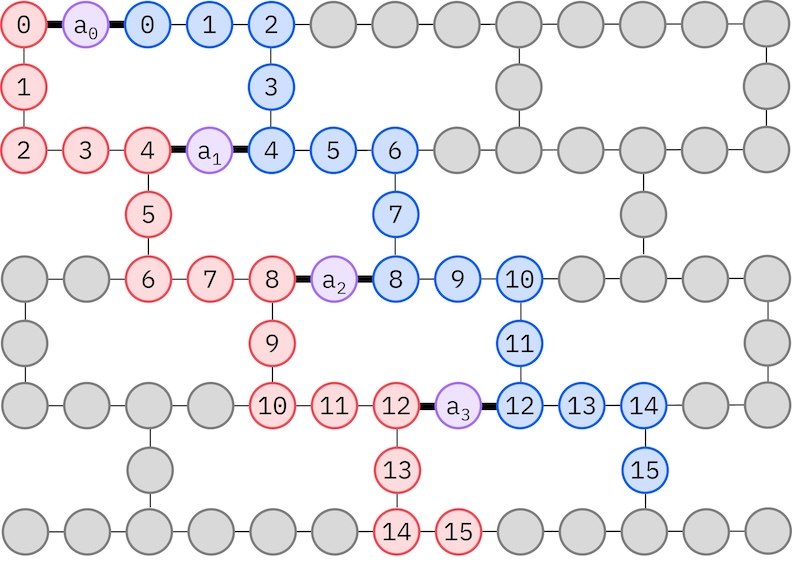
Deoarece hardware-ul IBM țintă are o topologie heavy-hex, vom adopta modelul zig-zag pentru interacțiunile dintre Qubiți. În acest model, orbitalele (reprezentate de Qubiți) cu același spin sunt conectate printr-o topologie liniară (cercuri roșii și albastre) unde fiecare linie ia o formă zig-zag din cauza conectivității heavy-hex a hardware-ului țintă. Tot din cauza topologiei heavy-hex, orbitalele pentru spinuri diferite au conexiuni la fiecare al 4-lea orbital (0, 4, 8 etc.) (cercuri violet).
import ffsim
from qiskit import QuantumCircuit, QuantumRegister
n_reps = 1
alpha_alpha_indices = [(p, p + 1) for p in range(num_orbitals - 1)]
alpha_beta_indices = [(p, p) for p in range(0, num_orbitals, 4)]
ucj_op = ffsim.UCJOpSpinBalanced.from_t_amplitudes(
t2=t2,
t1=t1,
n_reps=n_reps,
interaction_pairs=(alpha_alpha_indices, alpha_beta_indices),
)
nelec = (num_elec_a, num_elec_b)
# create an empty quantum circuit
qubits = QuantumRegister(2 * num_orbitals, name="q")
circuit = QuantumCircuit(qubits)
# prepare Hartree-Fock state as the reference state and append it to the quantum circuit
circuit.append(ffsim.qiskit.PrepareHartreeFockJW(num_orbitals, nelec), qubits)
# apply the UCJ operator to the reference state
circuit.append(ffsim.qiskit.UCJOpSpinBalancedJW(ucj_op), qubits)
circuit.measure_all()
Pasul 2: Optimizarea problemei
Acum vom optimiza Circuitul nostru pentru hardware-ul țintă. Trebuie să alegem dispozitivul hardware de utilizat înainte de a ne optimiza Circuitul. Vom folosi un Backend fals de 127 de Qubiți din qiskit_ibm_runtime pentru a emula un dispozitiv real. Pentru a rula pe un QPU real, înlocuiește pur și simplu Backend-ul fals cu unul real. Consultă documentația Qiskit IBM Runtime pentru mai multe informații.
from qiskit_ibm_runtime.fake_provider import FakeSherbrooke
backend = FakeSherbrooke()
Apoi, recomandăm următorii pași pentru a optimiza ansatz-ul și a-l face compatibil cu hardware-ul.
- Selectează Qubiți fizici (
initial_layout) de pe hardware-ul țintă care respectă modelul zig-zag descris mai sus. Dispunerea Qubiților în acest model duce la un circuit eficient, compatibil cu hardware-ul și cu mai puține Gate-uri. - Generează un pass manager pe etape folosind funcția generate_preset_pass_manager din Qiskit cu alegerea ta de
backendșiinitial_layout. - Setează etapa
pre_inita pass manager-ului tău pe etape laffsim.qiskit.PRE_INIT.ffsim.qiskit.PRE_INITinclude pase de Transpiler Qiskit care descompun Gate-urile în rotații orbitale și apoi le îmbină, rezultând mai puține Gate-uri în Circuitul final. - Rulează pass manager-ul pe Circuitul tău.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
spin_a_layout = [0, 14, 18, 19, 20, 33, 39, 40, 41, 53, 60, 61, 62, 72, 81, 82]
spin_b_layout = [2, 3, 4, 15, 22, 23, 24, 34, 43, 44, 45, 54, 64, 65, 66, 73]
initial_layout = spin_a_layout + spin_b_layout
pass_manager = generate_preset_pass_manager(
optimization_level=3, backend=backend, initial_layout=initial_layout
)
# without PRE_INIT passes
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/o pre-init passes): {isa_circuit.count_ops()}")
# with PRE_INIT passes
# We will use the circuit generated by this pass manager for hardware execution
pass_manager.pre_init = ffsim.qiskit.PRE_INIT
isa_circuit = pass_manager.run(circuit)
print(f"Gate counts (w/ pre-init passes): {isa_circuit.count_ops()}")
Gate counts (w/o pre-init passes): OrderedDict({'rz': 4420, 'sx': 3432, 'ecr': 1366, 'x': 239, 'measure': 32, 'barrier': 1})
Gate counts (w/ pre-init passes): OrderedDict({'rz': 2460, 'sx': 2156, 'ecr': 730, 'x': 71, 'measure': 32, 'barrier': 1})
Pasul 3: Executarea experimentelor
După optimizarea circuitului pentru execuția pe hardware, suntem pregătiți să îl rulăm pe hardware-ul țintă și să colectăm mostre pentru estimarea energiei stării fundamentale. Deoarece avem un singur circuit, vom folosi modul de execuție Job din Qiskit Runtime și vom executa circuitul nostru.
Notă: Am comentat codul pentru rularea circuitului pe un QPU și l-am lăsat ca referință pentru utilizator. În loc să rulăm pe hardware real în acest ghid, vom genera mostre aleatoare extrase din distribuția uniformă.
import numpy as np
from qiskit_addon_sqd.counts import generate_bit_array_uniform
# from qiskit_ibm_runtime import SamplerV2 as Sampler
# sampler = Sampler(mode=backend)
# job = sampler.run([isa_circuit], shots=10_000)
# primitive_result = job.result()
# pub_result = primitive_result[0]
# bit_array = pub_result.data.meas
rng = np.random.default_rng(24)
bit_array = generate_bit_array_uniform(10_000, num_orbitals * 2, rand_seed=rng)
Pasul 4: Post-procesarea rezultatelor
Acum rulăm algoritmul SQD folosind funcția diagonalize_fermionic_hamiltonian. Consultă documentația API pentru explicații privind argumentele acestei funcții.
Solverul inclus în addon-ul SQD folosește implementarea CI selectiv din PySCF, mai precis pyscf.fci.selected_ci.kernel_fixed_space. Exemplul de mai jos arată, de asemenea, cum să transmiți argumente cu cuvinte cheie acelei funcții prin intermediul solverului inclus. Aici transmitem argumentul max_cycle.
from functools import partial
from qiskit_addon_sqd.fermion import SCIResult, diagonalize_fermionic_hamiltonian, solve_sci_batch
# SQD options
energy_tol = 1e-3
occupancies_tol = 1e-3
max_iterations = 5
# Eigenstate solver options
num_batches = 1
samples_per_batch = 300
symmetrize_spin = True
carryover_threshold = 1e-4
max_cycle = 200
# Pass options to the built-in eigensolver. If you just want to use the defaults,
# you can omit this step, in which case you would not specify the sci_solver argument
# in the call to diagonalize_fermionic_hamiltonian below.
sci_solver = partial(solve_sci_batch, spin_sq=0.0, max_cycle=max_cycle)
# List to capture intermediate results
result_history = []
def callback(results: list[SCIResult]):
result_history.append(results)
iteration = len(result_history)
print(f"Iteration {iteration}")
for i, result in enumerate(results):
print(f"\tSubsample {i}")
print(f"\t\tEnergy: {result.energy + nuclear_repulsion_energy}")
print(f"\t\tSubspace dimension: {np.prod(result.sci_state.amplitudes.shape)}")
result = diagonalize_fermionic_hamiltonian(
hcore,
eri,
bit_array,
samples_per_batch=samples_per_batch,
norb=num_orbitals,
nelec=nelec,
num_batches=num_batches,
energy_tol=energy_tol,
occupancies_tol=occupancies_tol,
max_iterations=max_iterations,
sci_solver=sci_solver,
symmetrize_spin=symmetrize_spin,
carryover_threshold=carryover_threshold,
callback=callback,
seed=rng,
)
Iteration 1
Subsample 0
Energy: -105.45358671756313
Subspace dimension: 5476
Iteration 2
Subsample 0
Energy: -107.95172900082163
Subspace dimension: 249001
Iteration 3
Subsample 0
Energy: -108.97460330369815
Subspace dimension: 339889
Iteration 4
Subsample 0
Energy: -109.02739376648793
Subspace dimension: 440896
Iteration 5
Subsample 0
Energy: -109.030972328451
Subspace dimension: 597529
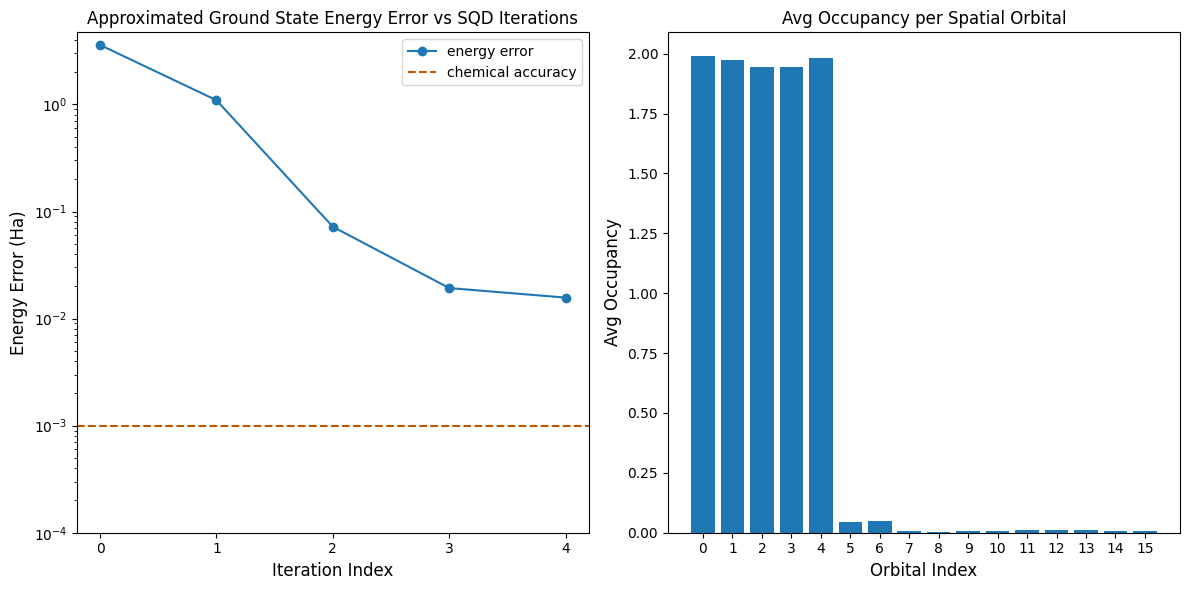
Acum reprezentăm grafic rezultatele.
Primul grafic arată că, după câteva iterații, estimăm energia stării fundamentale cu o eroare de aproximativ ~16 mH (acuratețea chimică este în general acceptată ca fiind 1 kcal/mol 1.6 mH). Ține minte că mostrele cuantice din această demonstrație au fost zgomot pur. Semnalul provine din cunoștințele a priori despre structura electronică și Hamiltonianul molecular.
Al doilea grafic arată ocupanța medie a fiecărei orbitale spațiale după iterația finală. Putem observa că atât electronii cu spin-up, cât și cei cu spin-down ocupă primele cinci orbitale cu probabilitate ridicată în soluțiile noastre.
import matplotlib.pyplot as plt
# Data for energies plot
x1 = range(len(result_history))
min_e = [
min(result, key=lambda res: res.energy).energy + nuclear_repulsion_energy
for result in result_history
]
e_diff = [abs(e - exact_energy) for e in min_e]
yt1 = [1.0, 1e-1, 1e-2, 1e-3, 1e-4]
# Chemical accuracy (+/- 1 milli-Hartree)
chem_accuracy = 0.001
# Data for avg spatial orbital occupancy
y2 = np.sum(result.orbital_occupancies, axis=0)
x2 = range(len(y2))
fig, axs = plt.subplots(1, 2, figsize=(12, 6))
# Plot energies
axs[0].plot(x1, e_diff, label="energy error", marker="o")
axs[0].set_xticks(x1)
axs[0].set_xticklabels(x1)
axs[0].set_yticks(yt1)
axs[0].set_yticklabels(yt1)
axs[0].set_yscale("log")
axs[0].set_ylim(1e-4)
axs[0].axhline(y=chem_accuracy, color="#BF5700", linestyle="--", label="chemical accuracy")
axs[0].set_title("Approximated Ground State Energy Error vs SQD Iterations")
axs[0].set_xlabel("Iteration Index", fontdict={"fontsize": 12})
axs[0].set_ylabel("Energy Error (Ha)", fontdict={"fontsize": 12})
axs[0].legend()
# Plot orbital occupancy
axs[1].bar(x2, y2, width=0.8)
axs[1].set_xticks(x2)
axs[1].set_xticklabels(x2)
axs[1].set_title("Avg Occupancy per Spatial Orbital")
axs[1].set_xlabel("Orbital Index", fontdict={"fontsize": 12})
axs[1].set_ylabel("Avg Occupancy", fontdict={"fontsize": 12})
print(f"Exact energy: {exact_energy:.5f} Ha")
print(f"SQD energy: {min_e[-1]:.5f} Ha")
print(f"Absolute error: {e_diff[-1]:.5f} Ha")
plt.tight_layout()
plt.show()
Exact energy: -109.04667 Ha
SQD energy: -109.03097 Ha
Absolute error: 0.01570 Ha