Variational Quantum Eigensolver (VQE)
Pentru acest modul, studenții trebuie să aibă un mediu Python funcțional și cele mai recente versiuni ale următoarelor pachete instalate:
qiskitqiskit_ibm_runtimeqiskit-aerqiskit.visualizationnumpypylatexenc
Pentru a configura și instala aceste pachete, consultă ghidul Instalare Qiskit. Pentru a rula joburi pe calculatoare cuantice reale, studenții vor trebui să creeze un cont IBM Cloud, urmând pașii din ghidul Configurarea contului tău IBM Cloud.
Acest modul a fost testat și a utilizat aproximativ 8 minute de timp QPU. Aceasta este o estimare, iar utilizarea ta reală poate varia.
# Added by doQumentation — required packages for this notebook
!pip install -q matplotlib numpy qiskit qiskit-aer qiskit-ibm-runtime scipy
# Uncomment and modify this line as needed to install dependencies
#!pip install 'qiskit>=2.1.0' 'qiskit-ibm-runtime>=0.40.1' 'qiskit-aer>=0.17.0' 'numpy' 'pylatexenc'
Introducere
De la dezvoltarea modelului cuantic în primele decenii ale secolului XX, oamenii de știință au înțeles că electronii nu urmează traiectorii fixe în jurul nucleului unui atom, ci există în regiuni de probabilitate numite orbitali. Acești orbitali corespund unor niveluri de energie specifice și discrete pe care electronii le pot ocupa. Electronii se găsesc în mod natural la cele mai joase niveluri de energie disponibile, cunoscute drept starea fundamentală. Totuși, dacă un electron absoarbe suficientă energie, poate sări la un nivel de energie superior, intrând într-o stare excitată. Această stare excitată este temporară, iar electronul va reveni în cele din urmă la un nivel de energie mai jos, eliberând energia absorbită, adesea sub formă de lumină. Acest proces fundamental de absorbție și emisie de energie este esențial pentru înțelegerea modului în care atomii interacționează și formează legături.
Când atomii se unesc pentru a forma molecule, orbitalii lor atomici se combină pentru a forma orbitali moleculari. Aranjamentul și nivelurile de energie ale electronilor din acești orbitali moleculari dictează proprietățile moleculei rezultate și rezistența legăturilor chimice. De exemplu, la formarea unei molecule de hidrogen () din doi atomi de hidrogen individuali, electronul din fiecare atom ocupă orbitali atomici. Pe măsură ce atomii se apropie, acești orbitali atomici se suprapun și se combină pentru a forma noi orbitali moleculari — unul cu energie mai mică (un orbital de legătură) și unul cu energie mai mare (un orbital anti-legătură). Cei doi electroni, câte unul din fiecare atom de hidrogen, vor ocupa preferențial orbitalul de legătură cu energie mai mică, conducând la formarea unei legături covalente stabile care menține împreună molecula . Diferența de energie dintre atomii separați și molecula formată, în special energia electronilor din orbitalii moleculari, determină stabilitatea și proprietățile legăturii.
În secțiunile următoare, vom explora acest proces de formare moleculară, concentrându-ne pe molecula . Vom folosi un calculator cuantic real, combinat cu tehnici de optimizare clasice, pentru a afla energia acestui proces simplu, dar fundamental. Acest experiment va oferi o demonstrație practică a modului în care calculul cuantic poate fi aplicat pentru a rezolva probleme de chimie computațională, oferind perspective asupra rolului energiei electronilor.
VQE - Un algoritm cuantic variațional pentru probleme de valori proprii
Tehnici de aproximare pentru chimie - principiul variațional și setul de bază
Contribuțiile lui Erwin Schrödinger la mecanica cuantică nu se limitează la introducerea unui nou model electronic; în esență, el a stabilit mecanica undelor prin dezvoltarea faimoasei ecuații Schrödinger dependente de timp:
Aici, este operatorul Hamiltonian, care reprezintă energia totală a sistemului, iar este funcția de undă ce conține toate informațiile despre starea cuantică a sistemului. (Notă: este derivata totală în raport cu timpul, iar valoarea proprie a energiei nu este inclusă explicit aici.)
Totuși, în multe aplicații practice — precum determinarea nivelurilor de energie permise ale atomilor și moleculelor — folosim în schimb ecuația Schrödinger independentă de timp (ecuația valorilor proprii ale energiei), care este derivată din forma dependentă de timp prin presupunerea unei stări staționare. O stare staționară este o stare cuantică în care densitatea de probabilitate de a găsi o particulă într-un punct dat din spațiu nu se schimbă în timp.
În această formă, reprezintă valoarea proprie a energiei corespunzătoare stării cuantice . Hamiltonianul include diverse contribuții energetice, cum ar fi energia cinetică a electronilor și nucleelor, forțele de atracție dintre electroni și nuclee, și forțele de respingere dintre electroni.
Rezolvarea ecuației valorilor proprii ale energiei ne permite să calculăm nivelurile de energie cuantificate ale sistemelor atomice și moleculare. Totuși, pentru molecule, rezolvarea ei exactă este dificilă deoarece funcția de undă , care descrie distribuția spațială a electronilor, este complexă și de înaltă dimensionalitate.
Prin urmare, oamenii de știință folosesc tehnici de aproximare pentru a obține soluții practice și precise. În această lucrare, ne vom concentra pe două metode cheie:
-
Principiul variațional
Această metodă aproximează funcția de undă și o ajustează pentru a se apropia cât mai mult posibil de energia țintă, de obicei energia stării fundamentale a sistemului. Ideea de bază din spatele principiului variațional este simplă:
- Dacă ghicim o funcție de undă (o „funcție de încercare"), energia calculată din ea va fi întotdeauna egală cu sau mai mare decât energia stării fundamentale () a sistemului.
- Ajustând parametrii din funcția de încercare, , putem obține o aproximare din ce în ce mai bună a energiei stării fundamentale.
- Precizia sa depinde în mare măsură de alegerea funcției de undă de încercare . O funcție de încercare aleasă greșit poate duce la o estimare a energiei departe de valoarea reală.
-
Aproximarea setului de bază
A doua metodă de aproximare intervine în etapa de construire a funcției de undă — abordarea prin set de bază. În chimia cuantică, rezolvarea exactă a ecuației Schrödinger pentru molecule este aproape imposibilă. În schimb, aproximăm funcția de undă complexă, cu mai mulți electroni, construind-o din funcții matematice mai simple, predefinite. Un set de bază este, în esență, o colecție de astfel de funcții matematice cunoscute, centrate de obicei pe atomii din moleculă, care sunt folosite ca blocuri de construcție pentru a reprezenta forma și comportamentul electronilor în sistem. Gândește-te la asta ca și cum ai încerca să recreezi o sculptură detaliată folosind doar o colecție de piese LEGO standard — cu cât ai mai multe tipuri și dimensiuni de piese (cu cât setul de bază este mai mare), cu atât poți aproxima mai fidel forma originală.
Aceste funcții de bază sunt adesea inspirate de soluțiile analitice pentru sisteme simple, cum ar fi atomul de hidrogen, luând forme precum funcții de tip Gaussian sau Slater, deși sunt în continuare aproximări. În loc să lucrăm cu orbitalii moleculari completi, teoretic „exacți", dar intractabili, îi exprimăm ca o combinație liniară (o sumă cu coeficienți) a acestor funcții de bază. Această metodă este cunoscută sub numele de abordarea Combinației Liniare a Orbitalilor Atomici (LCAO) atunci când funcțiile de bază seamănă cu orbitalii atomici. Prin optimizarea coeficienților din această combinație liniară, putem găsi cea mai bună funcție de undă aproximativă și energie posibile în limitele setului de bază ales.
- Cu cât sunt incluse mai multe funcții în setul de bază, cu atât aproximarea este mai bună, dar acest lucru vine cu prețul unui efort computațional mai mare.
- Un set de bază mic oferă o estimare aproximativă, în timp ce un set de bază mare oferă rezultate mai precise, cu prețul necesității unor resurse computaționale mai mari.
Pentru a rezuma, pentru a face calculele fezabile și a reduce costul computațional, folosim principiul variațional prin aproximarea funcției de undă, ceea ce reduce complexitatea computațională și permite optimizarea iterativă pentru minimizarea energiei. Între timp, abordarea prin set de bază simplifică calculele prin reprezentarea orbitalilor atomici ca o combinație de funcții predefinite, în loc să rezolve direct o funcție de undă continuă.
Verifică-ți înțelegerea
Consideră funcția de undă de încercare unde este o constantă de normalizare și este un parametru ajustabil.
(a) Normalizează funcția de undă de încercare determinând astfel încât .
Answer
Pentru a normaliza funcția de undă de încercare dată:
Folosește integrala Gaussiană:
setează , apoi obții:
(b) Calculează valoarea de așteptare a Hamiltonianului dat de unde , care corespunde unui potențial de oscilator armonic simplu.
Answer
Hamiltonianul pentru un oscilator armonic este:
Valoarea de așteptare a energiei cinetice
Calculând derivata a doua:
Astfel:
Folosind rezultatele standard ale integralelor Gaussiene:
Valoarea de așteptare a energiei potențiale
Folosind:
obținem:
Valoarea de așteptare a energiei totale
(c) Folosește principiul variațional pentru a găsi optim minimizând .
Answer
Optimizează pentru energia minimă
Diferențiază
Rezolvând:
Substituind în :
care corespunde exact energiei stării fundamentale a oscilatorului cuantic armonic.
VQE (Variational Quantum Eigensolver)
Variational quantum eigensolver (VQE) este metoda principală pe care o vom folosi pentru a explora procesul și, aici, vom analiza ce este VQE și cum funcționează. Dar să facem mai întâi o pauză și să privim un lucru foarte important prin întrebarea de verificare.
Verifică-ți înțelegerea
Dacă avem deja atât de multe strategii pentru problemele de chimie, de ce avem nevoie de un calculator cuantic? Și care este scopul folosirii atât a calculatoarelor cuantice, cât și a celor clasice împreună?
Answer
Calculul cuantic are șansa de a revoluționa chimia prin abordarea problemelor cu care calculatoarele clasice se confruntă din cauza scalării exponențiale a stărilor cuantice. Richard Feynman a remarcat în mod celebru că pentru a simula natura, calculele trebuie să fie și ele cuantice [ref 1].
De exemplu, simularea cafeinei cu cel mai simplu set de baze (STO-3G) ar necesita biți, mult mai mult decât numărul total de stele din universul observabil () [ref 2]. Un calculator cuantic poate descrie orbitalele electronice ale cafeinei cu 160 de Qubiți.
Calculatoarele cuantice procesează în mod natural interacțiunile cuantice folosind superpozița și entanglementul, care oferă o modalitate promițătoare de a permite simulări moleculare precise. Mai mult, putem combina avantajele atât ale calculatoarelor cuantice (simularea electronilor), cât și ale calculatoarelor clasice (pre/post-procesarea datelor, gestionarea proceselor algoritmice, optimizarea și altele). Acestea sunt așteptate să îmbunătățească descoperirea materialelor, proiectarea medicamentelor și predicțiile de reacție, reducând experimentele costisitoare de tip trial-and-error. [ref 3][ref 4]
Dacă vrei să știi de ce sunt necesare calculatoarele cuantice pentru problemele de chimie și de ce să folosești atât resurse de calcul cuantice, cât și clasice, consultă următoarele articole:
Acum să revenim la VQE.
VQE combină puterea calculatoarelor cuantice cu cele clasice, folosind în mod fundamental principii variaționale pentru a obține energia stării fundamentale a sistemului. Pentru a înțelege VQE, descompune-l mai întâi în trei părți:
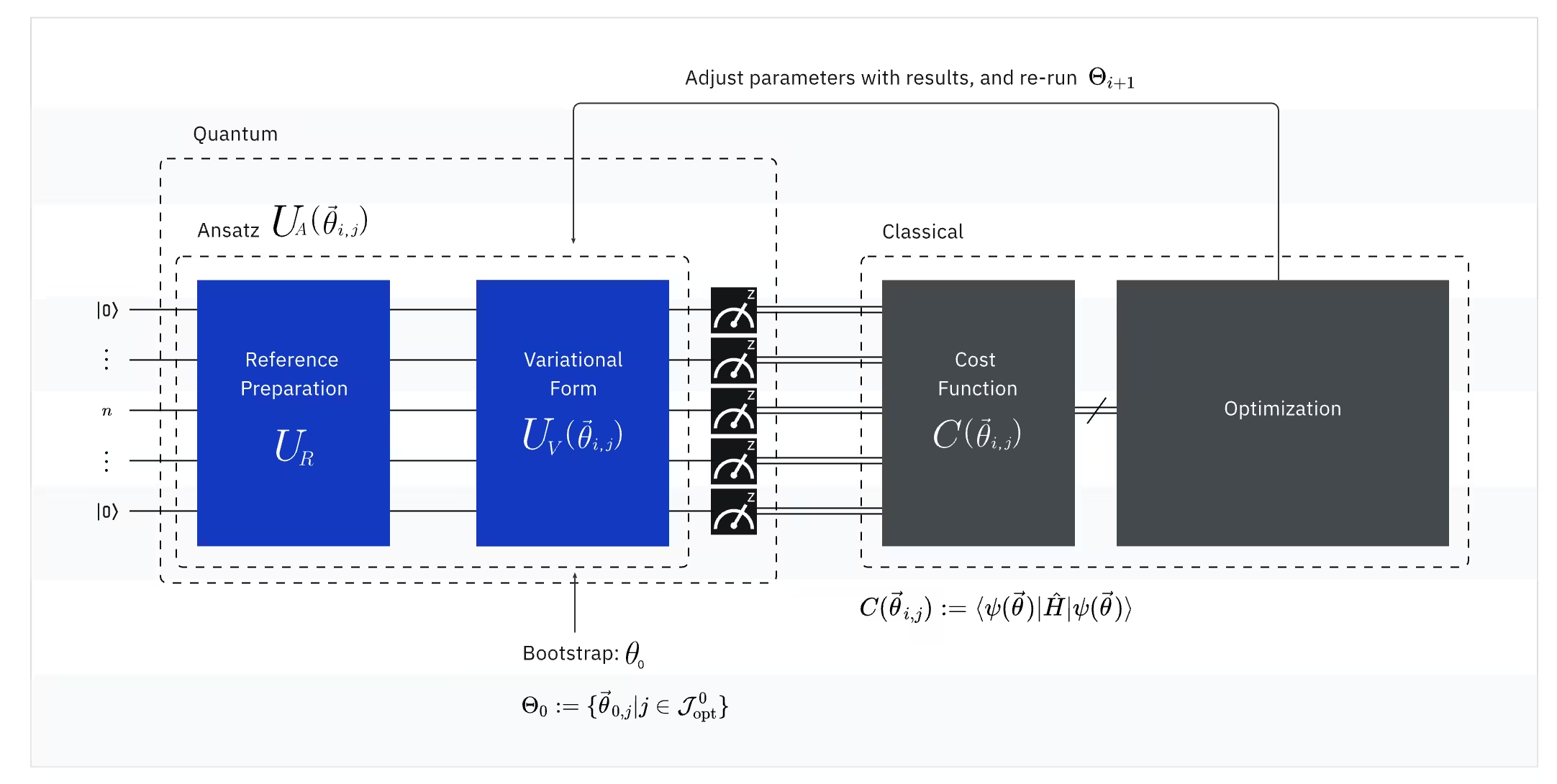
(Cuantic) Observable: Hamiltonianul molecular (energia unei molecule)
În VQE, Hamiltonianul molecular/atomic este un observable, ceea ce înseamnă că putem măsura valoarea sa printr-un experiment. Scopul nostru este să găsim cea mai mică energie posibilă (energia stării fundamentale) a moleculei. Pentru a face acest lucru, folosim o stare cuantică de testare, generată de un circuit cuantic parametrizat (ansatz). Măsurăm observabilul și optimizăm starea cuantică până atingem cea mai mică energie posibilă.
Setul de baze folosit pentru Hamiltonianul molecular determină numărul de Qubiți necesari și afectează direct precizia VQE. Alegerea setului de baze potrivit este esențială pentru echilibrarea eficienței și preciziei. Pentru a simplifica calculele fără a schimba setul de baze, putem folosi strategii precum impunerea simetriei și reducerea spațiului activ. Multe molecule au forme simetrice (precum un fluture sau un fulg de nea), ceea ce înseamnă că unele părți se comportă la fel. În loc să calculăm totul separat, ne putem concentra doar pe părțile unice, economisind resurse cuantice, valorificând astfel simetria. În reducerea spațiului activ, luăm în considerare doar orbitalii importanți, deoarece nu toți electronii influențează semnificativ energia moleculară. Electronii apropiați de nucleu rămân în mare parte nemodificați, în timp ce alții influențează legăturile. Prin aplicarea acestor metode, putem face VQE mai eficient, menținând în același timp precizia.
Odată ce obținem un Hamiltonian molecular folosind setul de baze adecvat și strategiile de mai sus, trebuie să transformăm acest Hamiltonian în unul potrivit pentru calculatoarele cuantice. Maparea problemelor la operatori Pauli poate fi destul de complicată. Acest lucru este valabil mai ales în chimia cuantică, care lucrează cu particule indistincte (electroni), deoarece Qubiții sunt distincți. Nu vom intra în detaliile mapărilor aici, dar te referim la următoarele resurse. O discuție generală despre maparea unei probleme la operatori cuantici poate fi găsită în Quantum computing in practice. O discuție mai detaliată despre maparea problemelor de chimie în operatori cuantici poate fi găsită în Quantum chemistry with VQE.
Pentru acest modul, îți vom furniza Hamiltonianele (cu un singur qubit) adecvate pentru și , astfel încât să ne putem concentra pe utilizarea calculatorului cuantic. Aceste Hamiltoniene cu un singur qubit sunt pregătite folosind setul de baze STO-6G și maparea Jordan-Wigner, care este maparea cea mai directă cu cea mai simplă interpretare fizică, deoarece mapează ocuparea unui spin-orbital la ocuparea unui qubit. De asemenea, am folosit o tehnică de reducere a Qubiților prin folosirea unei simetrii a Hamiltonianului, care utilizează tiparele din modul în care se comportă ocupațiile de spin pentru a reduce numărul de Qubiți. Pentru molecula , presupunem că distanța dintre cei doi atomi de hidrogen este 0.735 .
(Cuantic) Ansatz: Funcția de undă de testare (Cum să construiești o stare cuantică trivială cu un circuit cuantic)
Pentru VQE, ansatz-ul (plural: ansätze) constă din două componente cheie. Prima este pregătirea stării inițiale, care configurează starea Qubitului prin aplicarea de poartă-uri cuantice fără parametri variationali. A doua componentă este circuit-ul cuantic parametrizat, un circuit cuantic special cu parametri ajustabili, similar cu butoanele unui radio. Acești parametri vor fi folosiți pentru ultima parte — optimizatorul clasic — pentru a ne ajuta să atingem cea mai bună stare fundamentală posibilă.
În secțiunea despre principiul variational, am învățat că calitatea stării de testare afectează calitatea rezultatelor algoritmului variational. Aceasta înseamnă că alegerea unui ansatz bun este importantă în VQE. Încă o dată, acesta este un subiect bogat și complex. Nu vom acoperi diferitele tipuri de ansatz sau originile lor aici. Dacă ești interesat să afli mai multe despre Circuit-urile cuantice parametrizate și ansatz, poți explora lecția Ansatz and variational form din cursul de design al algoritmilor variationali, care oferă explicații detaliate și exemple de ansätze.
Deoarece vom folosi un Hamiltonian cu un singur qubit în acest modul, avem nevoie de un circuit cuantic parametrizat cu un singur qubit ca ansatz. Vom vedea trei tipuri de ansätze cu un singur qubit în secțiunea următoare. Le vom compara și vom discuta considerații cheie în selectarea unui ansatz.
(Clasic) Optimizer: ajustarea fină a Circuit-ului cuantic
Odată ce calculatorul cuantic măsoară energia observabilului din ansatz, parametrii ansatz-ului și valoarea energiei sunt trimise la optimizatorul clasic pentru ajustare. Acest proces de optimizare este efectuat pe un calculator clasic, folosind de obicei pachete științifice de uz general precum SciPy.
Optimizatorul clasic tratează energia măsurată ca o funcție de cost. În problemele de optimizare, o funcție de cost (numită uneori și funcție obiectiv) este o funcție matematică care măsoară cât de „bună" este o anumită soluție. Scopul optimizatorului este să găsească setul de parametri care minimizează această funcție de cost. În contextul găsirii energiei stării fundamentale a unei molecule, energia în sine servește ca funcție de cost — vrem să găsim parametrii pentru Circuit-ul nostru cuantic („soluția" noastră) care produc cea mai mică energie posibilă. Optimizatorul clasic folosește această valoare măsurată a energiei (costul) și determină următorul set de parametri optimizați pentru ansatz-ul cuantic. Acești parametri actualizați sunt apoi trimiși înapoi la circuit-ul cuantic, iar procesul se repetă. Cu fiecare iterație, optimizatorul clasic ajustează parametrii pentru a încerca să reducă energia (minimizând funcția de cost) până când este îndeplinit un criteriu de convergență predefinit, asigurând în mod ideal că este găsită cea mai mică energie posibilă (corespunzătoare stării fundamentale a moleculei pentru acea distanță de legătură și acel set de baze).
Există multe strategii de optimizare oferite de pachete științifice precum SciPy. Poți găsi mai multe în lecția Optimization loops din cursul Variational algorithm design. Aici vom folosi COBYLA (Constrained Optimization BY Linear Approximations), un algoritm de optimizare potrivit pentru peisaje energetice complicate. În special, COBYLA nu încearcă să calculeze un gradient al funcției studiate; acesta se numește un optimizer fără gradient. Imaginează-ți că încerci să găsești cel mai înalt vârf dintr-un lanț muntos cu ochii închiși. Deoarece nu poți vedea întregul peisaj, faci pași mici în diferite direcții, verificând dacă urci sau cobori. COBYLA funcționează într-un mod similar — se deplasează prin spațiul parametrilor, testând diferite valori, îmbunătățind treptat rezultatul până găsește cel mai bun.
Acum ești gata să efectuezi un calcul VQE. În acest scop, încearcă întrebarea de verificare de mai jos, care recapitulează procesul general.
Verifică-ți înțelegerea
Completează spațiile libere cu termenii corecți pentru a completa rezumatul procesului VQE, apoi dă click pentru a-ți verifica răspunsurile.
VQE este un algoritm cuantic variational, care combină puterea (1) ________ și a calculului clasic, folosit pentru a găsi (2) __________ a unei molecule. Procesul începe prin definirea (3) __________, care reprezintă energia totală a sistemului și acționează ca observable în măsurătorile cuantice. Apoi, pregătim un (4) __________, un circuit cuantic cu parametri ajustabili care reprezintă funcția de undă de testare a moleculei. Acești parametri sunt optimizați folosind un (5) __________, un algoritm clasic care ajustează parametrii iterativ pentru a minimiza energia măsurată. În discuția de mai sus, am folosit optimizatorul (6) __________, care rafinează parametrii ansatz-ului fără a necesita calcule de derivate. Procesul continuă până când atingem (7) __________, ceea ce înseamnă că am găsit cea mai mică energie posibilă a moleculei.
Bancă de cuvinte:
- classical optimizer
- ground state energy
- hardware-efficient
- ansatz
- molecular Hamiltonian
- COBYLA
- quantum computing
- convergence
Answer
1 → quantum computing
2 → ground state energy
3 → molecular Hamiltonian
4 → ansatz
5 → classical optimizer
6 → COBYLA
7 → convergence
Calculează energia stării fundamentale a unui atom de hidrogen cu VQE
Acum, hai să folosim ceea ce am învățat pentru a calcula energia stării fundamentale a unui atom de hidrogen. Pe parcursul modulului, vom folosi un cadru pentru calculul cuantic cunoscut sub numele de „Qiskit patterns" (tipare Qiskit), care împarte fluxurile de lucru în următorii pași:
- Pasul 1: Mapează datele de intrare clasice la o problemă cuantică
- Pasul 2: Optimizează problema pentru execuție cuantică
- Pasul 3: Execută folosind primitivele Qiskit Runtime
- Pasul 4: Post-procesare și analiză clasică

Vom urma în general acești pași.
Să începem prin încărcarea unor pachete necesare, inclusiv primitivele Qiskit Runtime. Vom selecta, de asemenea, cel mai puțin ocupat calculator cuantic disponibil.
Mai jos există cod pentru salvarea acreditărilor la prima utilizare. Asigură-te că ștergi aceste informații din notebook după ce le-ai salvat în mediul tău, astfel încât acreditările tale să nu fie distribuite accidental când distribui notebook-ul. Consultă Set up your IBM Cloud account și Initialize the service in an untrusted environment pentru mai multe îndrumări.
# Load the Qiskit Runtime service
from qiskit_ibm_runtime import QiskitRuntimeService
# Load the Runtime primitive and session
from qiskit_ibm_runtime import EstimatorV2 as Estimator
# Syntax for first saving your token. Delete these lines after saving your credentials.
# QiskitRuntimeService.save_account(channel='ibm_quantum_platform',
# instance = '<YOUR_IBM_INSTANCE_CRN>', token='<YOUR-API_KEY>', overwrite=True, set_as_default=True)
# service = QiskitRuntimeService(channel='ibm_quantum_platform')
# Load saved credentials
service = QiskitRuntimeService()
# Use the least busy backend, or uncomment the loading of a specific backend like "ibm_brisbane".
backend = service.least_busy(operational=True, simulator=False, min_num_qubits=127)
# backend = service.backend("ibm_brisbane")
print(backend.name)
ibm_brisbane
Celula de mai jos îți va permite să comutezi între utilizarea simulatorului sau a hardware-ului real pe parcursul notebook-ului. Îți recomandăm să o rulezi acum:
# Load the Aer simulator and generate a noise model based on the currently-selected backend.
from qiskit_aer import AerSimulator
from qiskit_aer.noise import NoiseModel
# Alternatively, load a fake backend with generic properties and define a simulator.
noise_model = NoiseModel.from_backend(backend)
# Define a simulator using Aer, and use it in Sampler.
backend_sim = AerSimulator(noise_model=noise_model)
Pasul 1: Mapează problema pe circuite și operatori cuantici
Începem calculul VQE definind Hamiltonianul pentru molecula de hidrogen () la o distanță specifică de legătură. Acest Hamiltonian reprezintă energia totală a sistemului în termeni de operatori pe qubit, fiind produs și mapat din sistemul molecular printr-o procedură standard: 1) utilizarea setului de baze STO-6G (o colecție specifică de funcții matematice folosite pentru a aproxima orbitalii electronici), 2) aplicarea mapării Jordan-Wigner (o tehnică de traducere a operatorilor fermionici ce descriu electronii în operatori pe qubit) și 3) reducerea Qubitului folosind paritatea Hamiltonianului pentru simplificarea problemei.
Așa cum am explicat anterior, energiile stării fundamentale calculate depind în mare măsură de alegerea setului de baze și de geometria moleculară (cum ar fi distanța de legătură). Pentru această configurație specifică și după aceste transformări, Hamiltonianul cuantic rezultat pe qubit este simplu:
Aici, reprezintă operatorul identitate, iar reprezintă operatorul Pauli-Z, acționând pe un singur qubit. Coeficienții sunt derivați din integralele calculate folosind setul de baze STO-6G la această distanță de legătură particulară cu transformarea corespunzătoare.
Cu acest Hamiltonian definit, putem folosi acum VQE pentru a calcula energia stării sale fundamentale. Este util să comparăm energia calculată a stării fundamentale cu valorile așteptate. Pentru un atom de hidrogen (H) izolat, energia stării fundamentale este exact -0,5 Hartree (în absența efectelor relativiste). Să calculăm energia exactă a stării fundamentale a Hamiltonianului nostru specific pe qubit, definit mai sus, și să o comparăm cu valorile cunoscute relevante.
from qiskit.quantum_info import SparsePauliOp
import numpy as np
# Qubit Hamiltonian of the hydrogen atom generated by using STO-3G basis set and parity mapping
Hamiltonian = SparsePauliOp.from_list([("I", -0.2355), ("Z", 0.2355)])
# exact ground state energy of Hamiltonian
A = np.array(Hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print(
"The exact ground state energy of the Hamiltonian is ",
min(eigenvalues).real,
"hartree",
)
h = min(eigenvalues.real)
The exact ground state energy of the Hamiltonian is -0.471 hartree
Apoi, avem nevoie de un circuit cuantic parametrizat, un ansatz, pentru a pregăti o funcție de undă de încercare pentru starea fundamentală. Scopul este de a găsi parametrii care minimizează valoarea de așteptare a energiei . Alegerea ansatz-ului este crucială, deoarece determină mulțimea stărilor cuantice posibile pe care le poate pregăti Circuit-ul nostru. Un ansatz „bun" este suficient de flexibil pentru a reprezenta o stare foarte apropiată de starea fundamentală reală a Hamiltonianului studiat, dar nu atât de complex încât să necesite prea mulți parametri sau un circuit prea profund pentru computerele cuantice actuale.
Aici, vom încerca trei ansatz-uri diferite pentru un singur qubit pentru a vedea care oferă o „acoperire" mai bună a stărilor cuantice posibile pe care le poate avea un singur qubit. Prin „acoperire" înțelegem gama de stări cuantice pe care le poate produce Circuit-ul ansatz prin varierea parametrilor săi.
Vom folosi trei ansatz-uri bazate pe combinații diferite de poartă-uri de rotație pentru un singur qubit:
- Ansatz cu un poartă de rotație pe un singur ax: Acest ansatz folosește rotații doar în jurul unui singur ax (). Pe sfera Bloch, aceasta corespunde deplasării doar de-a lungul unui cerc specific. Este cel mai puțin flexibil și acoperă un set limitat de stări.
- Două ansatz-uri cu Gate-uri de rotație pe doi axe: Aceste ansatz-uri combină rotații în jurul a doi axe diferite ( și ). Acest lucru ne permite să atingem o porțiune mai mare a sferei Bloch, față de rotația pe un singur ax.
Comparând rezultatele VQE obținute cu aceste trei ansatz-uri, putem vedea cum flexibilitatea și acoperirea spațiului de stări al ansatz-ului influențează capacitatea noastră de a găsi energia reală a stării fundamentale a Hamiltonianului nostru simplificat. Un ansatz mai flexibil are potențialul de a găsi o aproximare mai bună, dar ar putea fi și mai dificil pentru optimizatorul clasic.
from qiskit import QuantumCircuit
from qiskit.circuit import Parameter
from qiskit.quantum_info import Statevector, DensityMatrix, Pauli
theta = Parameter("θ")
phi = Parameter("φ")
lam = Parameter("λ")
ansatz1 = QuantumCircuit(1)
ansatz1.rx(theta, 0)
ansatz2 = QuantumCircuit(1)
ansatz2.rx(theta, 0)
ansatz2.rz(phi, 0)
ansatz3 = QuantumCircuit(1)
ansatz3.rx(theta, 0)
ansatz3.rz(phi, 0)
ansatz3.rx(lam, 0)
<qiskit.circuit.instructionset.InstructionSet at 0x1059def80>
Acum, să generăm 5000 de numere aleatoare pentru fiecare parametru și să reprezentăm grafic distribuția stărilor cuantice aleatoare, generate de cele trei ansatz-uri cu acești parametri aleatori. Te poți gândi la acești parametri ca la rotații în jurul unor axe diferite pe o suprafață sferică. Pentru a vedea distribuția stărilor cuantice, vom folosi sfera Bloch, o sferă tridimensională care arată starea unui singur qubit. Orice punct de pe sferă reprezintă o stare posibilă a Qubitului, unde polii de nord și de sud sunt ca „0" și „1" clasice, dar qubit-ul poate fi și oriunde între ele, demonstrând proprietăți cuantice speciale precum superpoziția. Mai întâi, pregătește funcțiile necesare pentru a reprezenta grafic sfera Bloch 3D și pentru a genera 5000 de parametri aleatori.
import matplotlib.pyplot as plt
def plot_bloch(bloch_vectors):
# Extract X, Y, Z coordinates for 3D projection
X_coords = bloch_vectors[:, 0]
Z_coords = bloch_vectors[:, 2]
# Compute Y coordinates from X and Z to approximate the full Bloch sphere projection
Y_coords = bloch_vectors[:, 1]
# Create 3D plot
fig = plt.figure(figsize=(8, 8))
ax = fig.add_subplot(111, projection="3d")
ax.scatter(X_coords, Y_coords, Z_coords, color="blue", alpha=0.6)
# Labels and title
ax.set_xlabel("X")
ax.set_ylabel("Y")
ax.set_zlabel("Z")
ax.set_title("Parameterized 1-Qubit Circuit on 3D Bloch Sphere")
# Set axis limits and make them equal
ax.set_xlim([-1, 1])
ax.set_ylim([-1, 1])
ax.set_zlim([-1, 1])
# Ensure equal aspect ratio for all axes
ax.set_box_aspect([1, 1, 1]) # Equal scaling for x, y, z axes
# Show grid
ax.grid(True)
plt.show()
num_samples = 5000 # Number of random states
theta_vals = np.random.uniform(0, 2 * np.pi, num_samples)
phi_vals = np.random.uniform(0, 2 * np.pi, num_samples)
lam_vals = np.random.uniform(0, 2 * np.pi, num_samples)
Să vedem cum funcționează primul nostru ansatz.
# List to store Bloch Sphere XZ coordinates
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create a circuit and bind parameters
qc = ansatz1
bound_qc = qc.assign_parameters({theta: theta_vals[i]}) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to a numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
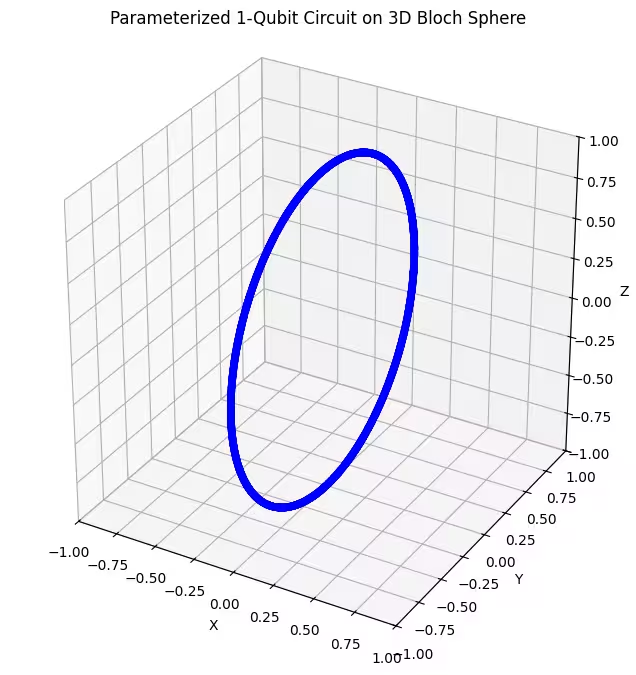
Putem observa că primul nostru ansatz returnează stări cuantice distribuite în formă de inel pe sfera Bloch. Acest lucru are sens, deoarece am dat ansatz-ului un singur parametru de rotație. Prin urmare, acesta poate produce doar stări rotite în jurul unui singur ax. Pornind din punctul și rotind în jurul unui singur ax, rezultatul va fi întotdeauna un inel. Să verificăm acum al doilea ansatz, care are două Gate-uri de rotație ortogonale - Rx și Rz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz2
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i]}
) # , lam: lam_vals[i]})
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
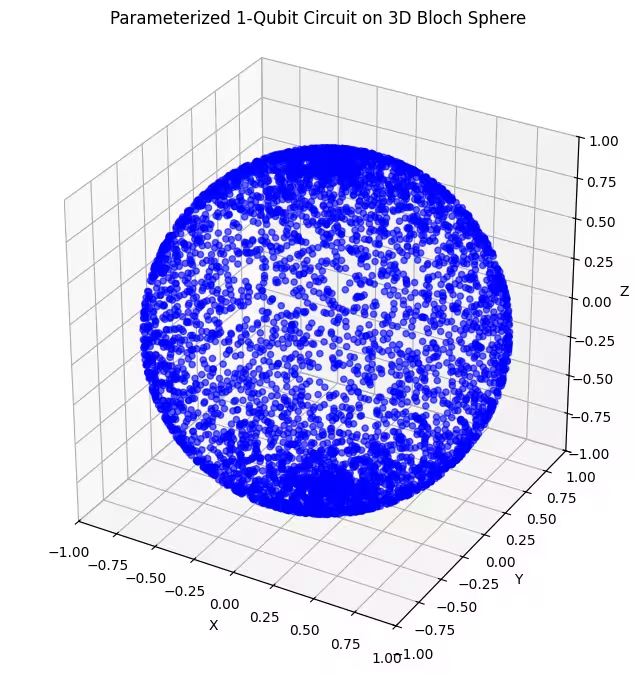
Aici putem vedea că al doilea ansatz acoperă o porțiune mai mare a sferei Bloch - dar observă că punctele sunt mai concentrate în jurul polilor și mai răspândite în jurul ecuatorului. Acum este momentul să verificăm ultimul nostru ansatz.
bloch_vectors = []
# Generate quantum states and extract Bloch vectors
for i in range(num_samples):
# Create circuit and bind parameters
qc = ansatz3
bound_qc = qc.assign_parameters(
{theta: theta_vals[i], phi: phi_vals[i], lam: lam_vals[i]}
)
state = Statevector.from_instruction(bound_qc)
rho = DensityMatrix(state)
X = rho.expectation_value(Pauli("X")).real
Y = rho.expectation_value(Pauli("Y")).real
Z = rho.expectation_value(Pauli("Z")).real
bloch_vectors.append([X, Y, Z]) # Store X, Z components
# Convert to numpy array for plotting
bloch_vectors = np.array(bloch_vectors)
plot_bloch(bloch_vectors)
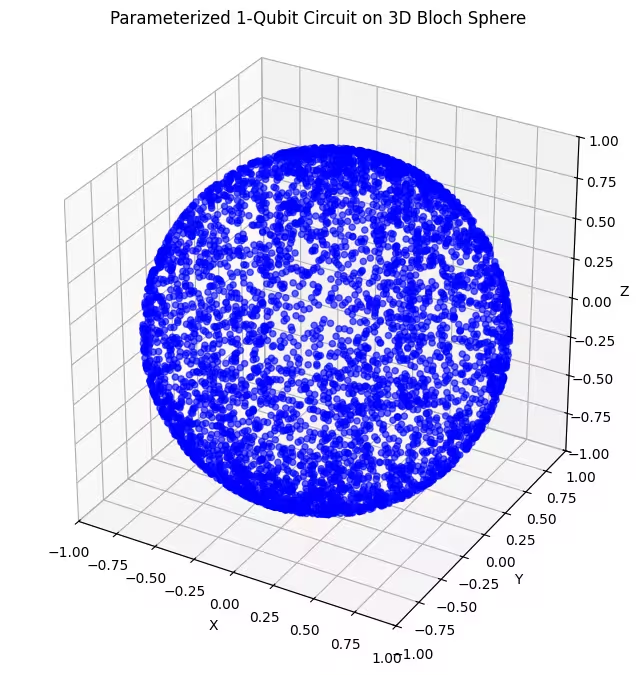
Aici poți vedea stări cuantice distribuite mai uniform, generate de ultimul nostru ansatz.
După cum s-a menționat, cel mai bun lucru de făcut este să obții cunoștințe despre starea fundamentală pe care o cauți și să folosești un ansatz bine potrivit pentru a sonda stări aproape de acea stare fundamentală. De exemplu, dacă am ști că starea noastră fundamentală se află lângă un pol, am putea selecta ansatz 2. Pentru simplitate, vom rămâne cu ansatz 3, care sondează uniform întreaga sferă Bloch.
Acum că am selectat ansatz-ul, să desenăm Circuit-ul.
# Pre-defined ansatz circuit and operator class for Hamiltonian
ansatz = ansatz3
num_params = ansatz.num_parameters
print("This circuit has ", num_params, "parameters")
ansatz.draw("mpl", style="iqp")
This circuit has 3 parameters

Pasul 2: Optimizează pentru hardware-ul țintă
Când rulăm un calcul pe un calculator cuantic real, nu ne interesează doar logica Circuit-ului cuantic. Ne interesează și lucruri precum ce operații poate efectua acel calculator cuantic particular și unde anume pe calculatorul cuantic se află qubit-urile pe care le folosim. Sunt unul lângă altul? Sunt departe unul de celălalt? Prin urmare, următorul pas este să rescriem Circuit-ul nostru folosind Gate-uri naturale pentru calculatorul cuantic pe care îl vom folosi, ținând cont și de dispunerea qubit-urilor. Acest lucru se poate face prin transpilation - după acest proces, poți vedea ansatz-ul nostru simplu convertit într-un set diferit de poartă-uri, iar qubit-urile noastre abstracte vor fi mapate pe qubit-uri fizice dintr-un calculator cuantic real.
from qiskit.transpiler.preset_passmanagers import generate_preset_pass_manager
config = backend.configuration()
print("Backend: {config.backend_name}")
print("Native gates: ", config.supported_instructions, ",")
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa = pm.run(ansatz)
ansatz_isa.draw(output="mpl", idle_wires=False, style="iqp")
Backend: {config.backend_name}
Native gates: ['ecr', 'id', 'delay', 'measure', 'reset', 'rz', 'sx', 'x'] ,

Poți observa că Gate-urile rx, rz ale ansatz-ului nostru au fost convertite într-o serie de poartă-uri rz, sx, care sunt Gate-urile native ale Backend-ului nostru. De asemenea, poți vedea că q0 este acum mapat pe al cincilea qubit fizic. Trebuie să mapăm și Hamiltonianul conform acestor modificări, ca în codul următor:
Hamiltonian_isa = Hamiltonian.apply_layout(layout=ansatz_isa.layout)
Pasul 3: Execuție pe hardware-ul țintă
A venit momentul să rulăm VQE-ul nostru pe un QPU real. Pentru aceasta, avem nevoie mai întâi de o funcție de cost pentru procesul de optimizare, care evaluează valoarea așteptată a Hamiltonianului cu o stare cuantică generată de ansatz. Nu-ți face griji! Nu trebuie să scrii totul de la zero. Am pregătit o funcție pentru asta, tot ce trebuie să faci este să rulezi celula de mai jos.
def cost_func(params, ansatz, hamiltonian, estimator):
"""Return estimate of energy from estimator
Parameters:
params (ndarray): Array of ansatz parameters
ansatz (QuantumCircuit): Parameterized ansatz circuit
hamiltonian (SparsePauliOp): Operator representation of Hamiltonian
estimator (EstimatorV2): Estimator primitive instance
cost_history_dict: Dictionary for storing intermediate results
Returns:
float: Energy estimate
"""
pub = (ansatz, [hamiltonian], [params])
result = estimator.run(pubs=[pub]).result()
energy = result[0].data.evs[0]
cost_history_dict["iters"] += 1
cost_history_dict["prev_vector"] = params
cost_history_dict["cost_history"].append(energy)
print(f"Iters. done: {cost_history_dict['iters']} [Current cost: {energy}]")
return energy
În final, pregătim parametrii inițiali pentru ansatz-ul nostru și procesul de optimizare al acestuia. Poți folosi pur și simplu toate valorile zero sau valori aleatoare. Am selectat parametrii inițiali mai jos, dar simte-te liber să comentezi sau să decomentezi linii din celulă pentru a eșantiona parametrii aleatoriu, uniform din 0 până la .
# x0 = np.random.uniform(0, 2*pi, 3)
x0 = [1, 1, 0]
# QPU Est. 2min for ibm_brisbane
from scipy.optimize import minimize
from qiskit_ibm_runtime import Batch
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, Hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 10, "tol": 0.01},
)
batch.close()
Iters. done: 1 [Current cost: -0.3361517318448143]
Iters. done: 2 [Current cost: -0.4682546422099432]
Iters. done: 3 [Current cost: -0.38985802144149584]
Iters. done: 4 [Current cost: -0.38319217316749354]
Iters. done: 5 [Current cost: -0.4628720756579032]
Iters. done: 6 [Current cost: -0.4683301936226905]
Iters. done: 7 [Current cost: -0.45480498699294747]
Iters. done: 8 [Current cost: -0.4690533242050814]
Iters. done: 9 [Current cost: -0.465867415110354]
Iters. done: 10 [Current cost: -0.4606882723137227]
h_vqe = res.fun
print("The reference ground state energy is ", min(eigenvalues))
print("The computed ground state energy is ", h_vqe)
The reference ground state energy is (-0.471+0j)
The computed ground state energy is -0.4690533242050814
Felicitări! Tocmai ai finalizat cu succes primul tău experiment de chimie cuantică. Putem observa o diferență între energia exactă a stării fundamentale a Hamiltonianului și cea calculată de noi, dar pentru că am folosit o tehnică implicită de atenuare a erorilor (care corectează erorile de citire), diferența este minoră. Este un start foarte bun!
Notă: Poți obține un rezultat mai bun setând un nivel de atenuare a erorilor folosind resilience_level. Valoarea implicită este 1, iar dacă setezi o valoare mai mare, va utiliza mai mult timp QPU, dar ar putea returna un rezultat mai bun.
Pasul 4: Post-procesare
A venit momentul să vedem cum a funcționat optimizatorul nostru clasic. Rulează celula de mai jos și observă tiparul de convergență.
fig, ax = plt.subplots()
x = np.linspace(0, 10, 10)
# Define the constant function
y_constant = np.full_like(x, h)
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Am pornit cu o valoare inițială destul de bună, astfel că am obținut o valoare finală bună în doar 10 pași. Poți vedea vârfuri mari și mici, aceasta fiind caracteristica tipică a optimizatorului COBYLA - acesta explorează spațiul de parcă nu poate vedea peisajul și ajustează dimensiunile pașilor cu fiecare măsurătoare.
Verifică-ți înțelegerea
Care este observația ta? Ce parte din procesul de mai sus poate fi îmbunătățită pentru a obține rezultate mai apropiate de valorile teoretice sau mai apropiate de energia precisă a stării fundamentale a Hamiltonianului? Ce aspecte trebuie luate în considerare?
Answer
Primul lucru de luat în considerare este schimbarea setului de baze folosite la calcularea Hamiltonianului moleculelor. Așa cum s-a menționat anterior, energia stării fundamentale a atomului H este -0,5 Hartree, după cum se știe, iar baza STO-6G pe care am ales-o nu este suficientă pentru a deriva cu precizie această valoare.
Alegerea unui tip de bază mai complex crește numărul de qubit-uri folosite de Hamiltonian; prin urmare, trebuie să selectăm un ansatz mai complex și mai potrivit pentru problemele de chimie.
Următorul aspect de optimizat este gestionarea zgomotului în QPU. Tehnicile mai avansate de atenuare a erorilor produc rezultate mai bune, dar pot dura mai mult. De asemenea, ia în considerare modul în care shot_number influențează rezultatele.
În final, o performanță de convergență mai bună poate fi obținută și prin încercarea unor optimizatori diferiți.
Calculează energia stării fundamentale a moleculei de hidrogen cu VQE
Acum că am parcurs procesul general al VQE folosind atomi , vom calcula mai rapid energia stării fundamentale a moleculei .
Pasul 1: Maparea problemei pe Circuit-uri cuantice și operatori
Îți punem la dispoziție și aici un Hamiltonian cu un singur qubit care folosește baza STO-6G și transformarea Jordan-Wigner, cu reducerea qubit-urilor prin utilizarea unei simetrii a Hamiltonianului. Rețineți că am folosit o distanță atomică între cei doi atomi de hidrogen de 0.735 .
Spre deosebire de calculul unui atom de hidrogen singular (), pentru a calcula starea fundamentală a unei molecule de hidrogen (), trebuie să luăm în considerare și forța repulsivă care acționează între nucleele celor doi atomi de hidrogen, pe lângă energia asociată orbitalilor electronici. În acest pas, vom da această valoare ca o constantă, iar în problema de verificare vom calcula efectiv această valoare.
h2_hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
# exact ground state energy of hamiltonian
nuclear_repulsion = 0.71997
A = np.array(h2_hamiltonian)
eigenvalues, eigenvectors = np.linalg.eig(A)
print("Electronic ground state energy (Hartree): ", min(eigenvalues).real)
print("Nuclear repulsion energy (Hartree): ", nuclear_repulsion)
print(
"Total ground state energy (Hartree): ", min(eigenvalues).real + nuclear_repulsion
)
h2 = min(eigenvalues).real + nuclear_repulsion
Electronic ground state energy (Hartree): -1.8659468547627318
Nuclear repulsion energy (Hartree): 0.71997
Total ground state energy (Hartree): -1.1459768547627318
Pasul 2: Optimizează pentru hardware-ul țintă
Deoarece numărul de qubiți folosiți de VQE și Hamiltonianul anterior este același cu cel al Backend-ului care va fi utilizat pentru execuție, vom folosi ansatz-ul existent și forma sa optimizată.
h2_hamiltonian_isa = h2_hamiltonian.apply_layout(layout=ansatz_isa.layout)
Pasul 3: Execută pe hardware-ul țintă
Acum este momentul să facem calculele pe QPU-ul real. Aproape totul este la fel, dar vom folosi punctul inițial corespunzător pentru a se potrivi cu Hamiltonianul. De asemenea, în partea iterativă, unele setări ale Estimator-ului, care este utilizat pentru a calcula valorile așteptate ale Hamiltonianului pentru ansatz pe QPU, vor fi configurate ușor diferit față de calculele anterioare. Vom discuta această modificare mai pe larg într-o întrebare de verificare.
x0 = [2, 0, 0]
# QPU time 4min for ibm_brisbane
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 10000
res = minimize(
cost_func,
x0,
args=(ansatz_isa, h2_hamiltonian_isa, estimator),
method="cobyla",
options={"maxiter": 15},
)
batch.close()
Iters. done: 1 [Current cost: -0.710621837568328]
Iters. done: 2 [Current cost: -0.2603208441168329]
Iters. done: 3 [Current cost: -0.25548711201326424]
Iters. done: 4 [Current cost: -0.581129450619904]
Iters. done: 5 [Current cost: -1.722920997605439]
Iters. done: 6 [Current cost: -1.6633324849371915]
Iters. done: 7 [Current cost: -1.8066989598929164]
Iters. done: 8 [Current cost: -1.8051093803839542]
Iters. done: 9 [Current cost: -1.802692217571555]
Iters. done: 10 [Current cost: -1.8233585485263144]
Iters. done: 11 [Current cost: -1.6904116652617205]
Iters. done: 12 [Current cost: -1.8245120321245392]
Iters. done: 13 [Current cost: -1.6837021361383608]
Iters. done: 14 [Current cost: -1.8166632606115467]
Iters. done: 15 [Current cost: -1.863446212658907]
h2_vqe = res.fun + nuclear_repulsion
print(
"The reference ground state energy is ", min(eigenvalues).real + nuclear_repulsion
)
print("The computed ground state energy is ", h2_vqe)
The reference ground state energy is -1.1459768547627318
The computed ground state energy is -1.143476212658907
Deși VQE oferă teoretic o limită superioară pentru energia reală a stării fundamentale, implementările practice pe hardware cuantic real sau simulat cu zgomot, precum și aproximările făcute la pregătirea Hamiltonianului (cum ar fi seturile de baze sau reducerea numărului de qubiți), pot introduce erori care uneori duc la o energie măsurată ușor mai mică decât valoarea teoretică exactă sau o valoare numerică de referință specifică. Deși există unele erori, rezultatele par satisfăcătoare, mai ales ținând cont de numărul mic de pași. Acum, hai să finalizăm acest calcul VQE analizând modul în care a funcționat optimizatorul.
Pasul 4: Post-procesare
fig, ax = plt.subplots()
x = np.linspace(0, 5, 15)
# Define the constant function
y_constant = np.full_like(x, min(eigenvalues))
ax.plot(
range(cost_history_dict["iters"]), cost_history_dict["cost_history"], label="VQE"
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
ax.plot(y_constant, label="Target")
plt.legend()
plt.draw()

Verifică-ți înțelegerea
Hai să calculăm energia de repulsie nucleară a moleculei , pe care am inclus-o ca valoare constantă (0.71997 Hartree).

Te rog folosește legea Coulomb și unitățile atomice pentru a te asigura că obții valoarea în Hartree.
Answer
Deoarece ambii nuclei de hidrogen sunt încărcați pozitiv, ei se resping reciproc prin forța electrostatică. Această respingere este descrisă de legea lui Coulomb:
unde este sarcina protonului, este permitivitatea vidului, iar este distanța dintre cei doi nuclei, măsurată în metri sau raze Bohr în unități de jouli (J).
Pentru a calcula această energie în Hartree, trebuie să convertim ecuația de mai sus în sistemul de Unități Atomice (AU). În AU, , , iar raza Bohr () este 1 și devine scara fundamentală de lungime în AU. Cu aceste simplificări, legea lui Coulomb se reduce la:
unde trebuie măsurat în raze Bohr ().
Pentru a converti separarea nucleară dată în în , avem nevoie de relația de conversie:
deci devine .
Prin urmare, energia de repulsie nucleară a unui dat este
Calculează energia de reacție a
Acum să folosim ceea ce am obținut! Ai utilizat VQE, un eigenresolvator cuantic variațional, pentru a calcula energia stării de bază a atomului și a moleculei . Ce mai rămâne este să folosim valorile calculate pentru a obține energia de reacție a procesului .
Energia de reacție este schimbarea de energie care are loc atunci când substanțele reacționează pentru a forma substanțe noi. Imaginează-ți că construiești ceva: uneori trebuie să introduci energie (cum ar fi stivuirea blocurilor), iar alteori energia este eliberată (ca o minge care coboară un deal). În chimie, reacțiile fie absorb energie (endotermice), fie eliberează energie (exotermice).
Energia de reacție a procesului poate fi calculată cu ajutorul formulei următoare:
Rulând celula de mai jos, hai să vedem asta vizual. Vom folosi valoarea exactă a stării de bază a fiecărui Hamiltonian și vom compara energia de reacție a soluției exacte cu rezultatele VQE.
# Theoretical values
E_H_theo = h.real
E_H2_theo = h2
# Experimental values
E_H_exp = h_vqe
E_H2_exp = h2_vqe
# Calculate reaction energies
E_reaction_theo = E_H2_theo - (2 * E_H_theo)
E_reaction_exp = E_H2_exp - (2 * E_H_exp)
# Set up the plot
fig, ax = plt.subplots(figsize=(8, 6))
ax.set_xlim(0, 3)
ax.set_ylim(-1.16, -0.93) # Adjust y-axis range to highlight differences
ax.set_xticks([])
ax.set_ylabel("Energy (Hartree)")
ax.set_title("H + H → H₂ Reaction Energy Diagram")
# Plot theoretical energy levels
ax.hlines(
y=2 * E_H_theo, xmin=0.5, xmax=1.3, linewidth=2, color="r", label="2H (Exact)"
)
ax.hlines(y=E_H2_theo, xmin=1.3, xmax=2, linewidth=2, color="b", label="H₂ (Exact)")
# Plot experimental energy levels
ax.hlines(
y=2 * E_H_exp,
xmin=0.5,
xmax=1.5,
linewidth=2,
color="r",
linestyle="dashed",
label="2H (VQE)",
)
ax.hlines(
y=E_H2_exp,
xmin=1.5,
xmax=2.5,
linewidth=2,
color="b",
linestyle="dashed",
label="H₂ (VQE)",
)
# Add labels
ax.text(
1,
2 * E_H_theo,
f"2H: {2*E_H_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
2,
E_H2_theo,
f"H₂: {E_H2_theo:.4f}",
verticalalignment="top",
horizontalalignment="left",
)
ax.text(
1,
2 * E_H_exp,
f"2H_VQE: {2*E_H_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
ax.text(
2,
E_H2_exp,
f"H₂_VQE: {E_H2_exp:.4f}",
verticalalignment="bottom",
horizontalalignment="right",
)
# Add arrows for reaction energy with ΔE label in the middle
mid_y_theo = (2 * E_H_theo + E_H2_theo) / 2
mid_y_exp = (2 * E_H_exp + E_H2_exp) / 2
ax.annotate(
"",
xy=(1.3, E_H2_theo),
xytext=(1.3, 2 * E_H_theo),
arrowprops=dict(arrowstyle="<->", color="g"),
)
ax.text(
1.35, mid_y_theo, f"ΔE: {E_reaction_theo:.4f}", color="g", verticalalignment="top"
)
ax.annotate(
"",
xy=(1.5, E_H2_exp),
xytext=(1.5, 2 * E_H_exp),
arrowprops=dict(arrowstyle="<->", color="g", linestyle="dashed"),
)
ax.text(
1.55,
mid_y_exp,
f"ΔE_VQE: {E_reaction_exp:.4f}",
color="g",
verticalalignment="center",
)
# Add legend
ax.legend()
plt.show()

După cum se arată în figură, deși există anumite erori, energia exactă a stării de bază a Hamiltonienilor și energia de reacție calculată folosind rezultatele VQE sunt similare, apropiindu-se de -0,2 Hartree.
Trebuie menționat că energia de reacție a acestui proces are o valoare negativă, ceea ce înseamnă că energia este eliberată prin intermediul procesului, iar molecula rezultată are o energie mai mică decât doi atomi individuali. 6. Concluzie
Hai să rezumăm ce am învățat până acum.
Am analizat mai întâi două tehnici de aproximare importante necesare pentru rezolvarea problemelor de chimie cuantică: principiul variațional și alegerea setului de bază, ambele fundamentale pentru VQE. Am explorat principiul variațional manual, calculând energia stării de bază a oscilatorului armonic simplu.
Apoi, am explorat VQE, un algoritm utilizat pe scară largă pentru calculul energiei stării de bază a unui sistem cuantic. Am rulat cod pentru a calcula energiile stării de bază ale hidrogenului atomic () și ale moleculei de hidrogen (). În particular, am aflat că este necesar să obținem Hamiltonianul molecular adecvat pentru sistem și să îl transformăm într-o formă executabilă pe un calculator cuantic. Am văzut, de asemenea, că ansatz-ul, un circuit cuantic parametrizat, este necesar pentru a pregăti stări cuantice de încercare în cadrul VQE și am discutat importanța alegerii unei structuri de circuit ansatz adecvate. Am aflat, de asemenea, că VQE se bazează pe un proces iterativ de optimizare folosind un calculator clasic, ghidând Circuit-ul cuantic pentru a găsi starea de energie minimă, și am văzut cum converge procesul.
În final, am utilizat energiile stărilor de bază ale și obținute prin VQE pentru a calcula energia de reacție a procesului .
VQE este un algoritm cuantic puternic pentru termen scurt, dar este important să fii conștient de limitările sale. Performanța VQE depinde în mare măsură de alegerea ansatz-ului — găsirea unui ansatz care poate fi pregătit eficient și care poate reprezenta cu acuratețe starea de bază reală devine dificilă pentru molecule mai mari și mai complexe. În plus, hardware-ul cuantic actual este susceptibil la zgomot, ceea ce poate afecta acuratețea rezultatelor VQE, în special pentru Circuit-uri mai adânci sau cu un număr mai mare de Qubiți. În ciuda acestor provocări, VQE servește drept algoritm fundamental, iar cercetările în curs explorează metode variaționale mai sofisticate și tehnici de atenuare a erorilor pentru a extinde limitele a ceea ce este posibil în chimia cuantică pe calculatoare cuantice de termen scurt. De exemplu, algoritmi precum Sample-based Quantum Diagonalization (SQD) sunt în curs de dezvoltare, care valorifică eșantioane obținute din Circuit-uri cuantice combinate cu diagonalizare clasică într-un subspațiu pentru a îmbunătăți estimarea energiei și a aborda unele dintre limitările cu care se confruntă VQE, în special în ceea ce privește eficiența măsurătorilor și robustețea la zgomot.
Recapitulare și întrebări
Concepte cheie:
- Algoritmul cuantic variațional este o paradigmă de calcul în care un calculator clasic și un calculator cuantic lucrează împreună pentru a rezolva o problemă.
- În VQE, pornim de la un Hamiltonian al sistemului nostru și îl mapăm pe Qubiți pentru execuție pe calculatorul cuantic. Selectăm un circuit cuantic parametrizat, un ansatz, și efectuăm măsurători repetate, variind parametrii ansatz-ului, până se atinge cea mai mică valoare de energie. Căutarea prin spațiul parametrilor se realizează folosind un optimizer clasic. Pentru a obține rezultate bune, este necesar să selectăm un ansatz bun și un optimizer adecvat.
- Energia de reacție este schimbarea totală de energie într-o reacție chimică, determinată de diferența dintre energia reactanților și a produșilor.
Adevărat/Fals
- Principiul variațional afirmă că valoarea de așteptare a energiei pentru orice funcție de undă de încercare este întotdeauna mai mare sau egală cu energia adevărată a stării de bază.
- Un set de bază este o colecție de funcții utilizate pentru a aproxima funcțiile de undă cuantice.
- VQE este un algoritm cuantic folosit pentru a rezolva exact ecuația Schrödinger pentru un Hamiltonian dat.
- În VQE, un circuit cuantic parametrizat (un ansatz) este folosit pentru a pregăti funcții de undă de încercare.
- Alegerea optimizer-ului în VQE (de exemplu, COBYLA, SPSA sau ADAM) nu afectează calitatea rezultatului.
Estimator-ul din Qiskit este folosit pentru a calcula direct valorile de așteptare ale Hamiltonienilor în VQE.
Întrebări cu variante multiple:
- Care este scopul Hamiltonianului în VQE?
- A) Să genereze stări cuantice aleatorii
- B) Să determine energia stărilor cuantice
- C) Să optimizeze Circuit-urile cuantice
- D) Să creeze entanglement
- Care este obiectivul principal al algoritmului VQE?
- A) Să găsească energia stării de bază a unui Hamiltonian
- B) Să creeze entanglement între Qubiți
- C) Să efectueze căutarea Grover
- D) Să spargă criptarea RSA
- Câte stări cuantice sunt generate în acest notebook pentru a compara ansatz-ul?
- A) 100
- B) 1000
- C) 5000
- D) 10.000
- De ce este necesar un optimizer clasic în VQE?
- A) Pentru a efectua măsurători cuantice
- B) Pentru a actualiza parametrii ansatz-ului în vederea minimizării energiei
- C) Pentru a crea entanglement între Qubiți
- D) Pentru a genera aleatorie cuantică
- De ce este ansatz-ul proiectat să fie parametrizat?
- A) Pentru a permite pregătirea stărilor cuantice
- B) Pentru a permite explorarea unui spațiu larg de stări cuantice
- C) Pentru a reduce complexitatea Circuit-ului
- D) Pentru a măsura direct valorile proprii
- Care dintre următoarele afirmații este cea mai corectă cu privire la alegerea unui ansatz bun?
- A) Un ansatz trebuie să producă stări distribuite uniform pe sfera Bloch, altfel va eșua.
- B) Un ansatz ar trebui adaptat sistemului tău pentru a se asigura că poate genera stări apropiate de starea de bază.
- C) Un ansatz ar trebui să producă stări aleatorii folosind parametrii săi variațional.
- D) Un ansatz mai bun are întotdeauna mai mulți parametri variațional.
(Opțional) Anexă: Suprasarcina optimizatorului în funcție de complexitatea ansatz-ului
VQE se confruntă cu câteva provocări bine cunoscute[ref 6], dintre care următoarele sunt legate de ceea ce am învățat mai sus.
- Provocările alegerii ansatz-ului
Există o provocare inerentă în alegerea ansatz-ului variațional potrivit. Ansatz-urile inspirate din chimie (cum ar fi UCCSD) oferă acuratețe fizică, dar necesită circuite adânci, în timp ce ansatz-urile eficiente hardware au circuite mai puțin adânci, dar pot lipsi de interpretabilitate fizică. De asemenea, multe ansatz-uri introduc parametri variațonali excesivi care contribuie puțin la îmbunătățirea acurateței, dar cresc semnificativ dificultatea optimizării.
- Dificultăți de optimizare
Peisajul optimizării VQE poate conține regiuni în care gradienții dispar exponențial (platouri sterpe), ceea ce face dificilă actualizarea eficientă a parametrilor variațonali de către optimizatorii clasici. Din această cauză, cercetătorii au încercat să utilizeze diferite tipuri de optimizatori — bazați pe gradient și fără gradient — însă ambii se confruntă cu provocări. Optimizatorii bazați pe gradient suferă din cauza platourilor sterpe, iar metodele fără gradient necesită un număr mare de evaluări ale funcției.
- Suprasarcina optimizatorului
O altă provocare bine-cunoscută este suprasarcina optimizatorului, care este legată de dimensiunea problemei. Circuitele cuantice necesare pentru VQE cresc în adâncime și complexitate pe măsură ce dimensiunea problemei crește; de obicei, aceasta implică și creșterea numărului de parametri de optimizat. Procesul de optimizare devine inextricabil pe măsură ce numărul parametrilor crește, ducând la convergență lentă și dificultăți în găsirea soluției optime.
Aici vom examina aceste provocări utilizând VQE pentru o moleculă , cu două tipuri diferite de ansatz-uri.
(Notă: Aceasta poate consuma mai mult timp QPU, deci nu ezita să folosești un simulator dacă nu ai suficient timp.)
from qiskit.circuit import ParameterVector
num_iter = 4
alpha = ParameterVector("alpha", 3)
beta = ParameterVector("beta", 3 * num_iter)
# step1: Map problem to quantum circuits and operators
hamiltonian = SparsePauliOp.from_list(
[("I", -1.04886087), ("Z", -0.7967368), ("X", 0.18121804)]
)
ansatz_1 = ansatz3
ansatz_2 = QuantumCircuit(1)
for i in range(num_iter):
ansatz_2.rx(beta[i * 3 + 0], 0)
ansatz_2.rz(beta[i * 3 + 1], 0)
ansatz_2.rx(beta[i * 3 + 2], 0)
ansatz_1.draw("mpl")
ansatz_2.draw("mpl")

# Step 2: Optimize for target hardware
target = backend.target
pm = generate_preset_pass_manager(target=target, optimization_level=3)
ansatz_isa_1 = pm.run(ansatz_1)
ansatz_isa_2 = pm.run(ansatz_2)
hamiltonian_isa_1 = hamiltonian.apply_layout(layout=ansatz_isa_1.layout)
hamiltonian_isa_2 = hamiltonian.apply_layout(layout=ansatz_isa_2.layout)
Acum să rulăm un VQE cu un punct inițial format din unuri, cu maximum 20 de pași, și să comparăm convergența celor două rulări.
# QPU time 3m 40s for ibm_brisbane
# Step 3: Execute on target hardware
from scipy.optimize import minimize
x0 = np.ones(ansatz_1.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_1, hamiltonian_isa_1, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.8782202668652658]
Iters. done: 2 [Current cost: -0.43473160695469165]
Iters. done: 3 [Current cost: -0.4076372093159749]
Iters. done: 4 [Current cost: -1.3587839859772106]
Iters. done: 5 [Current cost: -1.774529906754082]
Iters. done: 6 [Current cost: -1.541934983115727]
Iters. done: 7 [Current cost: -1.2732403113465345]
Iters. done: 8 [Current cost: -1.820842221085785]
Iters. done: 9 [Current cost: -1.8065762857059005]
Iters. done: 10 [Current cost: -1.8126394095981146]
Iters. done: 11 [Current cost: -1.8205831886180421]
Iters. done: 12 [Current cost: -1.8086715778994924]
Iters. done: 13 [Current cost: -1.8307676638629322]
Iters. done: 14 [Current cost: -1.8177328827556327]
Iters. done: 15 [Current cost: -1.8179426218088064]
Iters. done: 16 [Current cost: -1.8109239667991088]
Iters. done: 17 [Current cost: -1.824271872489647]
Iters. done: 18 [Current cost: -1.813167587671394]
Iters. done: 19 [Current cost: -1.824647343397313]
Iters. done: 20 [Current cost: -1.8219785311686143]
# Save Cost_history as a new list
ansatz_1_history = cost_history_dict["cost_history"]
# QPU time 3m 40s for ibm_brisbane
x0 = np.ones(ansatz_2.num_parameters)
batch = Batch(backend=backend)
cost_history_dict = {
"prev_vector": None,
"iters": 0,
"cost_history": [],
}
estimator = Estimator(mode=batch)
estimator.options.default_shots = 2048
res = minimize(
cost_func,
x0,
args=(ansatz_isa_2, hamiltonian_isa_2, estimator),
method="cobyla",
options={"maxiter": 20},
)
batch.close()
Iters. done: 1 [Current cost: -0.738191173881188]
Iters. done: 2 [Current cost: -0.42636037194506304]
Iters. done: 3 [Current cost: -1.3503788613797374]
Iters. done: 4 [Current cost: -0.9109204349776897]
Iters. done: 5 [Current cost: -0.9060873157510835]
Iters. done: 6 [Current cost: -0.7735065414083984]
Iters. done: 7 [Current cost: -1.586889197437709]
Iters. done: 8 [Current cost: -1.659215191584943]
Iters. done: 9 [Current cost: -1.245445981794618]
Iters. done: 10 [Current cost: -1.1608385766138023]
Iters. done: 11 [Current cost: -1.1551733876027737]
Iters. done: 12 [Current cost: -1.8143337768286332]
Iters. done: 13 [Current cost: -1.2510951563756598]
Iters. done: 14 [Current cost: -1.6918311531865413]
Iters. done: 15 [Current cost: -1.8163783305531838]
Iters. done: 16 [Current cost: -1.8434877732947152]
Iters. done: 17 [Current cost: -1.8461898233304472]
Iters. done: 18 [Current cost: -1.0346471214915485]
Iters. done: 19 [Current cost: -1.8322518854150687]
Iters. done: 20 [Current cost: -1.717144678705999]
ansatz_2_history = cost_history_dict["cost_history"]
fig, ax = plt.subplots()
# Define the constant function)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_1_history,
label="Ansatz with 3 parameters",
)
ax.plot(
range(cost_history_dict["iters"]),
ansatz_2_history,
label="Ansatz with 12 parameters",
)
ax.set_xlabel("Iterations")
ax.set_ylabel("Cost (Hartree)")
plt.legend()
plt.draw()

Graficul de mai sus demonstrează clar că procesul de optimizare al ansatz-ului cu mai multe variabile necesită mai mult timp pentru a ajunge la o convergență stabilă.
În loc să ne bazăm pe circuite simple cu un singur qubit și un ansatz direct, complexitatea optimizării crește atunci când sunt necesare circuite cuantice mai mari și ansatz-uri cu structuri mai complexe. Aceasta evidențiază o provocare bine-cunoscută în VQE: suprasarcina optimizatorului.
Cercetătorii continuă să dezvolte diverse metodologii avansate care pot folosi calculatoarele cuantice pentru probleme de chimie. Poți accesa o varietate de materiale educaționale la IBM Quantum Learning.
Referințe
- [ref 1 ] Richard P. Feynman, Simulating Physics with Computers, International Journal of Theoretical Physics, 1982.
- [ref 2] Marov, M.Y. (2015). The Structure of the Universe. In: The Fundamentals of Modern Astrophysics. Springer, New York, NY.
- [ref 3] How to solve difficult chemical engineering problems with quantum computing, IBM Research Blog, 2023.
- [ref 4] Y. Cao, J. Romero and A. Aspuru-Guzik, "Potential of quantum computing for drug discovery," in IBM Journal of Research and Development, vol. 62, no. 6, pp. 6:1-6:20, 1 Nov.-Dec. 2018
- [ref 5] Present State of Molecular Structure Calculation, REv. Mod. Phys. 32, 170, 1960
- [ref 6] Fedorov, D.A., Peng, B., Govind, N. et al. VQE method: a short survey and recent developments. Mater Theory 6, 2 (2022)