Mapare
Urmărește videoclipul despre mapare realizat de Olivia Lanes sau deschide videoclipul într-o fereastră separată pe YouTube.
Introducere
În această lecție, ne vom concentra pe primul și adesea cel mai dificil pas în definirea unui program cuantic: maparea unei probleme pentru a fi rulată pe un calculator cuantic. Acest pas acoperă modul în care un utilizator pornește de la o problemă computațională și o traduce în ceva ce poate fi rezolvat pe un calculator cuantic.
În Lecțiile 2 și 3 ale acestui curs, am menționat că etapa de mapare este prima din cele patru etape totale ale cadrului Qiskit patterns. Din aceste lecții, poate îți amintești că scopul mapării este să traducă sau să rescrie o problemă computațională într-o funcție de cost sau o valoare de așteptare pe care o putem evalua folosind un calculator cuantic.
În Lecția 3 am discutat un exemplu concret cu Max-Cut, o problemă computațional dificilă, dar foarte comună, în optimizarea combinatorie. În acel exemplu, am parcurs mai mulți pași pentru a traduce problema inițială de graf într-una care putea fi rezolvată pe un calculator cuantic. Am transformat problema găsirii numărului maxim de tăieturi în graf într-o funcție de cost, am rescris acea funcție de cost ca un Hamiltonian și am pregătit o stare cuantică de probă a cărei stare fundamentală corespundea tăieturii maxime. În final, am construit un circuit cuantic care reprezenta starea cuantică de probă de interes și am adăugat porțile specifice pentru a permite stării să evolueze în timp. Această secvență de pași a făcut parte din mapare. Deși pașii exacți au fost unici pentru problema Max-Cut, aceeași procedură generală poate fi aplicată multor alte aplicații, cum ar fi chimia cuantică și simulările cuantice.
Maparea poate fi dificilă. Nu există o strategie universală pentru fiecare problemă în parte, deci poate fi intimidantă. În această lecție, vom analiza câteva considerații generale pentru mapare și apoi vom intra în detaliile unor exemple reprezentative pentru a demonstra diferitele moduri de a mapa o problemă pe un calculator cuantic.
Considerații generale
Deși strategia exactă pe care o folosești pentru a mapa o problemă pe un calculator cuantic depinde de problema în cauză, în general aceasta îndeplinește câteva lucruri principale.
În primul rând, este posibil să trebuiești să simplifici problema pentru a o face fezabilă. Acest lucru nu este unic pentru calculul cuantic – toate disciplinele științifice folosesc modele simplificate pentru a studia fenomenele care le interesează, ignorând detaliile irelevante. În fizică, există o expresie celebră care duce acest principiu la extrem: „presupune o vacă sferică." Adesea este prea dificil să descrii un sistem exact așa cum apare, dar putem face în schimb simplificări rezonabile care pot duce totuși la soluții utile. Câteva exemple despre cum am putea face asta în calculul cuantic sunt limitarea dimensiunii sau adâncimii circuitului prin alegerea unui ansatz eficient hardware, truncherea evoluțiilor de timp complexe sau neglijarea termenilor Hamiltonieni care contribuie puțin la energia finală a unei stări cuantice.
În al doilea rând, maparea implică scrierea problemei în așa fel încât un calculator cuantic să o poată înțelege. Adesea, asta presupune să răspunzi la aceste trei întrebări:
- Ce vor reprezenta qubiții noștri în modelul nostru?
- Este problema noastră continuă? Deoarece calculatoarele cuantice sunt digitale, dacă problema este continuă, va trebui să găsim o modalitate de a o discretiza.
- În ultimul rând, topologia problemei se aliniază cu topologia hardware-ului? Dacă nu, ar putea fi nevoie să implementăm câteva trucuri pentru a o face să funcționeze.
Să abordăm prima întrebare, care este în centrul multora dintre dificultățile legate de înțelegerea mapării: Ce poate reprezenta un qubit?
Qubiții pot fi folosiți pentru a reprezenta multe lucruri. Primul, și poate cel mai simplu, este un nod dintr-un graf. Grafurile sunt folosite pentru a arăta conectivitatea în multe tipuri diferite de probleme matematice, iar nodurile sunt un element fundamental care reprezintă un punct sau o entitate într-o rețea. În funcție de ce reprezintă întreaga rețea, acesta ar putea fi un oraș, o persoană sau un ferromagnet într-o rețea cristalină.
Qubiții pot fi folosiți și pentru a reprezenta bosoni și fermioni, deși voi avertiza că un singur qubit nu este exact egal cu un boson sau un fermion – este puțin mai complicat decât atât, după cum vom discuta mai departe în lecție.
Acum ajungem la exemple puțin mai complicate. Pentru aceste modele, nu mai are sens să vorbim în termeni de qubiți singulari; în schimb, avem nevoie de grupuri de qubiți pentru a reprezenta ceva fizic. De exemplu, un grup de qubiți, reprezentat aici pe o topologie hexagonală grea, poate fi folosit pentru a reprezenta locații geometrice ale aminoacizilor; lanțuri polimerice. Un alt exemplu este simularea împrăștierii hadronice în modele de fizică a energiilor înalte, care poate fi realizată prin simularea evoluției în timp a unui pachet de undă hadronic. În acest caz, un registru de qubiți poate fi folosit pentru a codifica diferite stări ale unui câmp cuantic; starea de vid a acelui câmp sau un pachet de undă care se propagă deasupra acelui vid.
Dar în acest moment am vorbit suficient de abstract despre provocarea din fața noastră. Să analizăm aceste exemple în detaliu.
Exemple de mapare
Max-Cut
Să începem cu primul nostru exemplu. Una dintre cele mai simple probleme de mapare este cea pe care am acoperit-o deja în detaliu: exemplul Max-Cut. În acea problemă, maparea a fost destul de ușoară pentru noi, deoarece un qubit era echivalent cu un nod din graful nostru.
Reamintește-ți că, pentru a mapa problema Max-Cut, am exprimat funcția de cost ca un Hamiltonian folosind formularea QUBO. O funcție de cost Hamiltonian este o funcție care codifică soluția optimă a problemei în starea fundamentală a Hamiltonianului. Pentru a construi Hamiltonianul de cost, am folosit clasa SparsePauliOp din Qiskit pentru a specifica conectivitatea grafului nostru, iar etapa de mapare la operatorii cuantici a fost finalizată. Iar circuitul cuantic a fost pur și simplu ansatz-ul QAOA. Pentru o recapitulare, consultă lecția QAOA la scară de utilitate, unde parcurgem toate acestea în mult mai mult detaliu.
În acea lecție, chiar și în exemplul de 100 de qubiți la scară de utilitate, conectivitatea grafului se potrivea deja cu topologia hardware-ului nostru superconductor. Deci nu a trebuit să ne preocupăm de cum să gestionăm topologii diferite. Dar nu este întotdeauna cazul. Dacă am fi avut un graf mai complicat sau mai dens conectat decât exemplele evidențiate până acum, ar fi trebuit să implementăm o serie de porți SWAP pentru a modifica conectivitatea efectivă a hardware-ului. Acest lucru este gestionat în al doilea pas al Qiskit patterns, transpilarea, dar ar trebui ținut minte chiar și în etapa de mapare.
Plierea proteinelor
Acum, hai să explorăm un exemplu modelat în articolul numit „Resource-efficient quantum algorithm for protein folding", scris de IBM® și colaboratori de la Universitatea din New South Wales.
Puțin context de biochimie: Proteinele sunt macromolecule compuse din lanțuri lungi de aminoacizi. Aceste lanțuri se pliau în structuri complexe care îndeplinesc o mare varietate de funcții biologice. Determinarea structurii unei proteine în spațiul tridimensional și înțelegerea relațiilor dintre structură și funcție sunt printre cele mai dificile probleme din biochimia actuală. Proteinele se pliau în structuri utile datorită interacțiunilor dintre aminoacizi. Pe măsură ce o structură se răsucește și se pliau, aminoacizii care sunt îndepărtați unii de alții de-a lungul lanțului pot ajunge să fie unul lângă altul și pot interacționa puternic.
Pentru a modela acest lucru pe un calculator cuantic, avem nevoie de un Hamiltonian care să descrie toate aceste interacțiuni dintre aminoacizi. Apoi, putem prezice structura finală găsind starea care va minimiza energia Hamiltonianului nostru. Aici, ne vom concentra pe modul în care lanțurile de aminoacizi pot fi modelate pe un calculator cuantic și pe modul în care putem obține distanțe inter-aminoacid pentru calculul energiilor de interacțiune. Astfel, vom fi adunat toate contribuțiile necesare la Hamiltonianul necesar pentru a-l simula pe un calculator cuantic.
În proteinele reale, aminoacizii pot ocupa un continuum de locații posibile. Cu toate acestea, vom folosi o simplificare și îi vom restrânge folosind un model de rețea, unde fiecare aminoacid ocupă un punct pe un grilaj. Aici, autorii au folosit o rețea tetraedrică. O scurtă notă: codificăm direcția muchiilor, nu nodurile în sine, ca în problema Max-Cut. Fiecare qubit reprezintă un posibil pas de-a lungul grilajului tetaedric. Rețineți că siturile adiacente au fost etichetate A sau B din cauza orientărilor lor diferite în rețea.
Lanțul proteic este reprezentat ca o serie de rotații sau direcții pe această rețea. Fiecare rotație dintre aminoacizi poate fi într-una din cele patru direcții, corespunzând muchiilor tetraedrului. Aceste patru rotații posibile sunt codificate folosind patru qubiți în stările 0001, 0010, 0100 sau 1000.
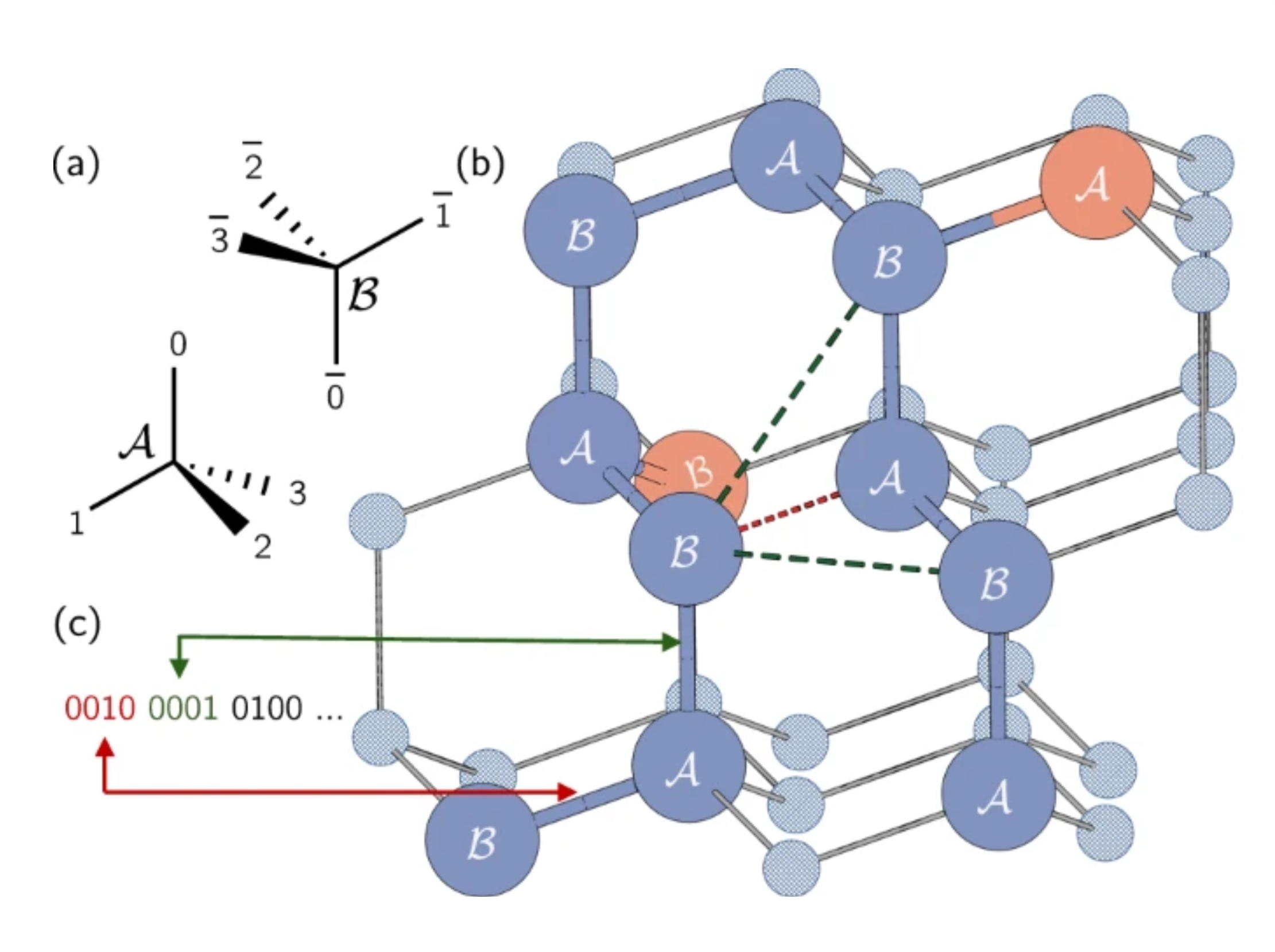
Să ne uităm la un exemplu în figura de mai sus. Să plasăm primul nostru aminoacid pe punctul etichetat „B", încercuit în roșu pe rețeaua noastră tetraedrică. Direcția de la primul aminoacid la al doilea este arbitrară, deoarece sistemul poate fi întotdeauna rotit pentru a face acea muchie să indice în orice direcție dorim. Prin urmare, putem plasa al doilea aminoacid pe punctul de dedesubtul primului, etichetat „A". Nu este la fel de ușor de observat, dar traseul de la al doilea la al treilea este, de asemenea, arbitrar. Toate cele trei alegeri ar rezulta în două muchii cu un unghi de aproximativ 109,5 grade între ele. Alegerea acestei a doua muchii determină pur și simplu orientarea proteinei noastre în spațiu. Prin urmare, fără pierdere de generalitate, putem alege ca primele două rotații să fie 0001 și 0010.
Cu aceste simplificări, configurația lanțului de aminoacizi este dată de această expresie:
Până acum, am mapat muchiile tetraedrului la qubiți, iar circuitul nostru cuantic va fi un ansatz. Acum trebuie să luăm în considerare modul de codificare a energiei problemei într-un Hamiltonian, astfel încât starea sa de bază să ne ofere modelul optim de pliere.
Pentru orice configurație dată, va exista o energie asociată datorată interacțiunilor dintre aminoacizi. Aceste interacțiuni sunt cele mai puternice atunci când cei doi aminoacizi sunt apropiați unul de celălalt. Evident, aminoacizii adiacenți în structura principală a lanțului vor interacționa întotdeauna între ei. Dar deoarece proteina se poate răsuci și plia, alte perechi de aminoacizi pot interacționa, de asemenea. Aminoacizii 10 și 20 s-ar putea ajunge pe situri adiacente după ce proteina s-a pliat, de exemplu. Prin urmare, avem nevoie de o formulă pentru a descrie distanța dintre aminoacizii și folosind informațiile codificate pe qubiții de configurație. Astfel putem folosi distanța lor de separare pentru a determina cât de puternic interacționează.
Mai întâi, hai să introducem o funcție care indică dacă o muchie este sau nu folosită pentru rotația la aminoacidul . Aici poate lua oricare dintre cele patru direcții posibile. Pe baza configurației cu care am început mai sus, putem scrie aceste funcții:
Putem defini apoi o diferență în numărul de rotații etichetate pe rețelele A și B de la indicele la indicele ca :
De ce am face asta? Ei bine, se dovedește că o rotație de la situl de rețea A la B este exact anulată de o rotație de la situl de rețea B la A. Deci, pentru a cunoaște distanța aminoacidului de pe situl față de cel de pe situl , trebuie doar să găsim diferența dintre pașii efectuați de-a lungul muchiilor de la siturile A și siturile B. Deoarece siturile A și B alternează în mod necesar de-a lungul structurii principale a proteinei, aceasta este captată în . Același argument se aplică tuturor celor patru tipuri de muchii. Astfel, distanța totală dintre aminoacizi în pași pe rețeaua tetraedrică poate fi calculată prin această expresie:
Dar cum obținem Hamiltonianul din această ecuație lungă pentru distanța totală dintre aminoacizi? Mai întâi, putem converti distanța în pași pe rețea în spațiul euclidian cu ajutorul unei geometrii simple:
Aceste distanțe vor fi folosite ulterior la calculul energiei configurației proteice. În funcție de scopurile noastre, am putea stabili o distanță limită sub care considerăm că perechea interacționează, sau am putea face ceva mai complex.
S-ar putea să nu fie evident, dar prin aceasta am terminat de fapt etapa de mapare. Stările qubiților indică „rotația" proteinei la fiecare sit de rețea, iar colecția de rotații determină distanța dintre orice pereche de aminoacizi. Perechile de aminoacizi din specii diferite au interacțiuni diferite — unele atractive, altele repulsive. Dacă folosești configurația și distanțele doar pentru a determina dacă interacțiunile cunoscute dintre aminoacizi sunt „active" sau „inactive", intensitățile acestora au fost deja calculate și pot fi pur și simplu consultate într-un tabel ca acesta:
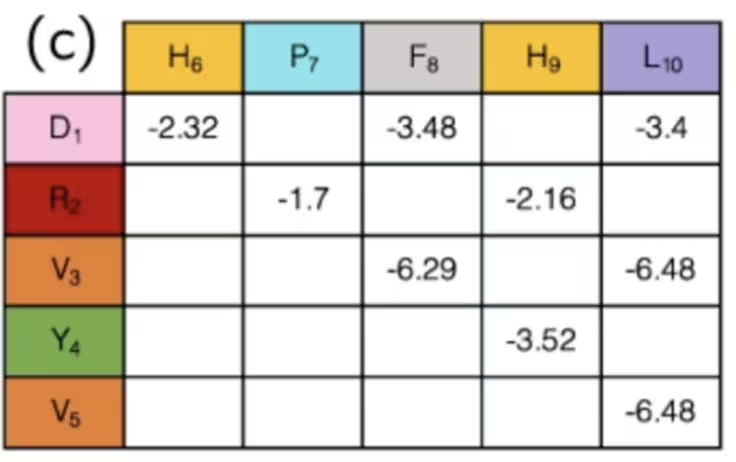
În concluzie, în acest exemplu, qubiții sunt folosiți pentru a marca pași pe un traseu de-a lungul unei rețele, care, împreună, formează un lanț de aminoacizi. Simulând modul în care se îndoaie și se pliau, putem spera să găsim rezultate mai bune în cercetarea medicală. Am omis calculul câtorva termeni ai acestui Hamiltonian deoarece erau extrem de specifici acestei probleme, în timp ce definirea direcțiilor pe o rețea poate fi aplicată mai general. Odată ce ai un Hamiltonian general, vei dori întotdeauna să îl traduci în operatori Pauli, care sunt nativi calculatorului cuantic. Despre asta vom discuta în continuare.
Transformarea Jordan-Wigner
Acum haideți să explorăm cum se traduce un sistem de particule subatomice în operatori Pauli.
Particulele subatomice sunt împărțite în două categorii – bosoni și fermioni. Bosonii, precum fotonii sau bosonul Higgs, respectă un anumit set de reguli statistice. Fermionii, precum electronii sau neutrinii, respectă altul. Diferența esențială dintre ei este că bosonii pot ocupa aceeași stare – nu există nicio limită pentru câți bosoni pot fi în starea fundamentală sau în orice stare excitată. Fermionii, pe de altă parte, sunt egoiști – ei cer ca fiecare particulă să aibă propria stare cuantică.
Bosonii au, de asemenea, spin întreg, în timp ce fermioni au spin semi-întreg, precum electronul cu spin-1/2 și particule mai exotice cu spin-3/2. Pentru a descrie un sistem de particule, avem nevoie de o descriere a energiei lor. Să ne concentrăm pe fermioni. Putem începe cu un Hamiltonian scris în termenii a ceea ce numim operatori c. Aceștia sunt, practic, obiecte matematice care corespund creării sau anihilării unui fermion dintr-o stare din sistem. Aceștia sunt adesea scrisi ca și , unde este operatorul care creează un fermion în starea și este operatorul care distruge un fermion în starea .
Dar să nu uităm că, de obicei, calculatoarele cuantice operează într-o bază de qubiți cu reguli specifice pentru reprezentarea sistemelor fermionice; ele nu lucrează în mod inerent în limbajul operatorilor fermionici. Pentru a acoperi acest decalaj, trebuie să mapăm această notație fermionică pe operatori Pauli, care corespund în mod natural porților cuantice.
Există mai multe astfel de transformări pentru fermioni. O alegere frecventă este transformarea Jordan-Wigner. Mapările Bravyi-Kitaev și de paritate sunt codificări fermionice mai recente. Operatorii bosonici pot fi transformați folosind transformarea Holstein-Primakoff sau mapând direct stările Fock la o sub-bază a modurilor bosonice disponibile, printre alte opțiuni. Alte codificări sunt, de asemenea, cercetate activ. Deocamdată, vom aborda doar transformarea Jordan-Wigner.
Transformarea Jordan-Wigner presupune maparea unui singur fermion pe mai mulți qubiți. De ce nu putem pur și simplu atribui câte un qubit fiecărui electron? Acest lucru are legătură cu distingibilitatea fermionilor identici. Qubiții sunt distingibili, iar electronii nu sunt. De exemplu, putem eticheta și identifica cu ușurință qubiții individuali pe orice dispozitiv. Dar indistingibilitatea electronilor înseamnă că nu îi putem eticheta deloc. Astfel, trebuie să etichetăm de fapt operatorii în funcție de orbitalii ocupați, precum 1s, 2p, 2p etc., nu în funcție de electronii specifici. Prin urmare, fiecare qubit joacă aproximativ rolul unui orbital din moleculă, care este fie ocupat, fie neocupat. Dar modul în care facem asta este puțin mai complicat. Maparea Jordan-Wigner ține evidența anti-simetriei și asigură statisticile corecte ale sistemului fermionic global. Maparea Jordan-Wigner exprimă operatorii fermionici în termenii operatorilor Pauli folosind aceste relații:
Maparea Jordan-Wigner este conceptual simplă datorită corespondenței unu-la-unu dintre orbitali și qubiți. Există și alte mapări care ating un obiectiv similar, inclusiv maparea de paritate. Calculul parității unei stări necesită luarea în considerare a mai multor qubiți. În maparea de paritate (și în unele altele) interpretarea unui qubit corespunzând unui orbital nu mai este valabilă. Acum haideți să parcurgem un exemplu simplu. Să presupunem că dorim să calculăm interacțiunea cu un singur qubit . Începem prin a înlocui definițiile noastre pentru operatorii de creare și anihilare.
Comutatorul . Prin urmare, expresia finală devine:
Astfel, am rescris cu succes o expresie fermionică în termenii operatorilor Pauli pe care calculatorul nostru cuantic îi va putea înțelege.
Haideți să discutăm rapid cum am implementa maparea Jordan-Wigner în Qiskit. Este important să înțelegi cum funcționează aceste tipuri de transformări, dar ar fi impractică realizarea lor manuală de fiecare dată – mai ales pentru sisteme de dimensiuni mari. Din fericire, Qiskit ne simplifică această sarcină cu funcția SparsePauliOp.
La nivel înalt, pașii sunt:
- Folosești funcția
from_lista lui SparsePauliOp pentru a crea un operator identitate corespunzător dimensiunii spațiului de parametri care trebuie mapat. - Urmând definiția operatorilor de creare și distrugere prezentată anterior, folosești funcția
from_lista lui SparsePauliOp pentru a defini operatorii Pauli , , . Aceasta va mapa operatorii fermionici de creare și anihilare pe operatorii de spin ai qubitului, codificând numărul de ocupare fermionic în baza de calcul a qubiților. - Generezi Hamiltonianul dorit în baza Pauli aplicând operatorii de mai sus orbitalilor de interes. Aceasta corespunde de obicei creării unei matrice identitate care reprezintă orbitalii de bază (neinteracționanți) și apoi aplicării operatorilor de creare și anihilare la spațiul activ, cu ponderi corespunzătoare specificului problemei în cauză.
Acum că înțelegi pe deplin schema de mapare Jordan-Wigner, haideți să vedem un exemplu mai complicat. Poate îți amintești de articolul intitulat „Scalable Circuits for Preparing Ground States on Digital Quantum Computers: The Schwinger Model Vacuum on 100 Qubits" din lecția anterioară. Nu vom trece din nou prin articol în detaliu — ne vom concentra doar pe maparea Jordan-Wigner, care este folosită pentru a exprima Hamiltonianul siturilor de rețea cu situri pentru modelul Schwinger în absența unui câmp electric.
Aici, este mult mai greu să identifici cu precizie ce reprezintă un qubit în acest model, deoarece doar colecția de qubiți împreună formează ceva fizic, în acest caz, un pachet de unde. În schimb, poți considera aproximativ qubiții ca bucăți discretizate de spațiu. Aici, este volumul rețelei în care fiecare element (celulă unitară) cuprinde doi qubiți. Operatorii fermionici pe care i-am văzut în diapozitivul anterior descriu amplitudinea funcției de undă pe un anumit sit. Astfel, Hamiltonianul nostru conține acești operatori fermionici de creare și anihilare. Prin urmare, folosim transformarea Jordan-Wigner pentru a mapa acești operatori pe operatorii Pauli.
unde este operatorul Pauli și este Odată ce avem un Hamiltonian scris în acest format, partea dificilă a etapei de mapare s-a încheiat și acum poate fi scris cu ușurință într-un circuit în operatori Pauli.
Concluzie
Am discutat patru exemple despre cum au fost folosite recent tehnici specifice de mapare în domeniul calculului cuantic, pornind de la cele mai simple și ajungând la aplicarea transformării Jordan-Wigner la dinamica hadronilor. O mare parte din acest material a fost foarte tehnică și, dacă nu ai mai întâlnit-o înainte, poate părea foarte intimidantă. Dar devine mai ușor cu cât petreci mai mult timp practicând – de aceea acest curs se numește Calculul Cuantic în Practică. Nu este ceva ce oricine poate lua de la zero și aplica imediat – necesită studiu, multă gândire și probabil momente frustrante. Dar te încurajez să rămâi cu acel disconfort și să explorezi cu adevărat întrebările care apar pe măsură ce continui. Este singura modalitate de a învăța.